 |
| ||||||
|
| ||||||||
|
| ||||||||
Home  Publications Publications |
|
|
Note to the Reader Proc. Natl. Acad. Sci. USA Vol. 89, pp. 1184 - 1188, February 1992 Neurobiology
Print Friendly ABSTRACT: One of the most striking results of
recent cell-lineage studies of vertebrate retina is the marked variability
in the size and types of clones marked by retroviral transfection and dye
injection of embryonic progenitor cells. Is this variability due to
microenvironmental modulation of cell determination, to lineage restriction,
or to experimental perturbation of the progenitor cells? We have taken
advantage of species-specific DNA probes to mark groups of lineage-related
cells in experimental mouse chimeras. This method of marking cells has two
distinct advantages over previous methods: direct manipulation of progenitor
cells is avoided, and clones are established at an earlier stage of retinal
development. The most notable feature of retinal cohorts in chimeras is
their structural uniformity—each is a solid radial array that contains the
same ratio of major cell types as the retina itself. This is true even of
the smallest monoclonal cohorts, which contain fewer than 200 cells. Our
result provides compelling empirical support for the hypothesis that the
murine retina is made up of hundreds of relatively homogeneous radial units,
each derived from single retinal precursor cells. This finding is
inconsistent with microenvironmental modulation of clone structure early in
development. We raise the possibility that the heterogeneity among clones
marked by dye injection and transfection is due to progressive lineage
restriction or to experimental perturbation of the retinal progenitor cells. The vertebrate retina is made up of numerous highly
differentiated cell types that originate from a common sheet of progenitor
cells that buds out from the central nervous system early in development (1,
2). Recent studies have taken advantage of the accessibility and neat
laminar organization of vertebrate retina to explore lineage relations
between retinal cell types (3–6). An important finding of this work has been
that single clones of retinal cells contain as many as six cell types. Even
a small clone with fewer than 25 cells may include rods, bipolar cells, and
a few MŸller glia (3). A second key result, and the focus of the current
study, has been the finding that the cellular composition of clones is
remarkably variable. For example, the ratio between rods and bipolar cells
in single clones varies between 2:1 and 20:1 in a single animal (6). This is
true even when progenitors are labeled at the same stage of development and
even when the clones they generate are situated in the same part of the
retina. There are at least three possible reasons for this
remarkable variability.  (i)The retinal progenitors may
themselves be heterogeneous and generate highly variable, but perhaps
well-defined, ratios of cell types.  (ii)The retinal progenitors
may be homogeneous, but strong interactions between uncommitted progeny and
the retinal microenvironment may ultimately generate clonal diversity (3-6).
&NBSP(iii)The experi mental manipulations used to mark clones early
in development—whether retroviral infection or direct dye injection—may
perturb the progenitors and the subsequent differentia tion of their
progeny. Whatever the source, it is difficult to reconcile the extraordinary
variability between neighboring clones with the highly ordered retinal
mosaic and the well-defined ratios of retinal cell subtypes in vertebrate
retina (7). This regularity is now known to be present even early in retinal
development, before cells are postmitotic (8). In the present study we address the problem of the
clonal heterogeneity in the retina using experimental mouse chimeras. In
these mice, progenitor cells of two genotypes are intermixed in the
embryonic retina. Cells of both genotypes can be labeled using  in
situ probes that are complementary to satellite DNA sequences (9, 10).
This method has allowed us to study progeny relationships among cohorts of
cells, all of which have the same genotype. Our approach complements
retroviral and dye labeling methods. One advantage of this procedure is that
it does not involve direct manipulation of the retina or of individual
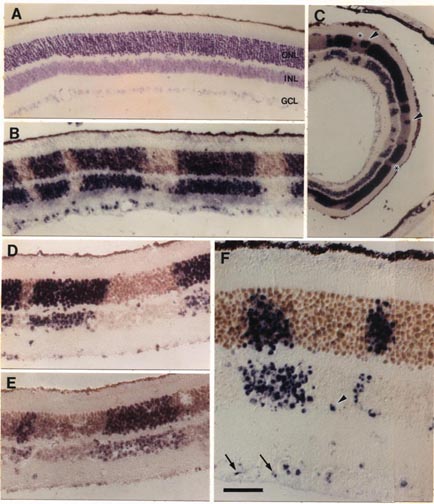
progenitor cells. Chimera Generation. Chimeric mice were made by combining 8-cell embryos of the domestic mouse  Mus musculus and the wild mouse  Mus caroli using conventional methods (10, 11). The fused blastocysts were placed into the uterine horns of pseudopregnant females. At maturity, chimeric mice were perfused with a 1:3 mixture (vol/vol) of acetic acid and ethanol. Eyes were hemisected and embedded in paraffin. Series of 5-Μm sections were cut from the retinal rim toward the optic nerve head. Consequently, the angle of section with respect to retinal layers changed gradually during cutting from radial to tangential. Series of sections were mounted on slides coated with 0.02% poly(L-lysine). Tissue was hybrid ized overnight at 56 °C in a solution containing a biotinylated dUTP-labeled DNA probe—either a M. caroli probe (pCR9) or a M. musculus probe (pMSat5)—in combination with 20% (wt/vol) dextran sulfate and 5x standard saline citrate. The in situ probes hybridize selectively with highly repetitive species-specific satellite DNA (ref. 12 and see Fig. 1). Hybridization sites were visualized using an avidin-alkaline phosphatase-catalyzed histochemical reaction.
The hybridization label was intense and selective. Single-labeled and unlabeled cells could be clearly identified. Using the two probes on adjacent sections, we consistently ob tained complementary labeling patterns (see Fig. 1 D and E). All retinal cell types, including glia, neurons, and photore ceptors, were well stained using either probe. Analysis. This study is based on the qualitative and quan titative examination of the distribution of cell genotypes in 20 retinas from 11 adult chimeras. The quantitative analysis (see Table 1 and Fig. 2) is based on a subset of small, isolated, and symmetrical cohorts such as those illustrated in Fig. 1B. As explained in more detail in Results, this sample is enriched in monoclonal cohorts. The positions of cohorts were plotted and their sizes were measured at x250. Cohort size was expressed as the number of cell diameters in a horizontal plane measured at the level of the outer nuclear layer. We found this parameter useful because it is insensitive to differential tissue shrinkage and case-to-case variation in cell size. Cohort size was usually expressed as a narrow range (e.g., three to four cell diameters). Whenever possible, serial sections were examined to determine the shape and maxi mum width of the cohort. Particular attention was taken to establish in these series whether cohorts extended fully and with even breadth through inner and outer nuclear layers. An estimate of cell number in each cohort was obtained by multiplying the square of its radius (r measured in cell diameters) by ir times the number of cell bodies intersected by a line running radially through the retina (retinal cell depth).
RESULTS
The eyes and neural retinas of chimeras are generally indis tinguishable from those of normal mice (Fig. lA). In both control and chimeric tissue, the area of the mouse retina is between 13 and 15 mm2, and the full thickness of the retina, including rod outer segments, averages °ree;°ree;150 um and 75 um in central and peripheral regions, respectively. In every chimeric retina, cells are clustered in sharply segregated cell cohorts of the same genotype (Fig. 1 B—ID. In contrast, intermingling of cells of both genotypes is common in the thalami of these animals. This makes it unlikely that the coherence of cohorts in the retina is due simply to adhesion among cells of the same genotype. Cohort boundaries are remarkably precise (Fig. 1) and can be defined with a precision of 5—10 um (one to two cell diameters). This precision is found in small symmetrical cohorts (Fig. 1B) and in large and often irregular shaped cohorts (Fig. iC). These boundaries are, with very few exceptions, oriented straight across the retinal layers. Al though this can often be difficult to appreciate in single sections, analysis of serial sections reveals that cohort ori entation is almost always radial. One clear exception to the rule of sharp segregation and radial orientation of cells of like genotype involves astrocytes and endothelial cells—cells that are not generated by the retinal neuroepithelium (13). These cells appear to be dis tributed without respect to the genotype of other cells. We also noted cells located along the margins of the inner and outer plexiform layers (e.g., Fig. iF) in regions populated principally by cells of the other genotype. These cells appear to be amacrine and horizontal cells. The ectopic cells are typically located within 50 um of cohorts of their own genotype. They may have migrated a short distance horizon tally during development. It may be relevant that amacrine and horizontal cells are the two retinal cell types most involved in the tangential integration of retinal activity. Uniformity of Cohorts. By tracing through serial sections, we found that the diameter of single cohorts is uniform through both inner and outer nuclear layers. The diameter of the set of smaller isolated cohorts that we could trace through serial sections varied by no more than one or two cell widths. In a sense, cohorts are core samples of the retina. This can be visualized well in Fig. 1B. In this fortuitous section, in which the section angle is almost perfectly radial, the uni formity of cohort diameter through the inner and outer nuclear layers is obvious and remarkable. However, in many other cases, such as shown in Fig. 1 C and F, cohorts appear to be tapered or even appear to be restricted to a single layer. Without exception, the analysis of serial sections demon strates that cohorts, whether large or small or whether well isolated or near other cohorts of the same genotype, extend as solid arrays of cells through the two nuclear layers. No fully reconstructed cohorts could be shown to be genuinely tapered. The tapered appearance is due to the angle between the axis of the cohort and the cutting angle. For example, in Fig. iF the larger cohort to the left of the photomicrograph has been cut through the middle of its inner nuclear layer component and through the edge of its outer nuclear layer component. In many cases, cohorts include one or more aligned retinal ganglion cells. However, the alignment of ganglion cells with cohorts in the nuclear layers is not always convincing (e.g., Fig. iF). This apparent lack of good alignment of ganglion cells may be due to the obliquity of section, to disruption or dispersion brought about by the normal and severe attrition of ganglion cells during development, or to horizontal shear between the ganglion cell layer and the nuclear layers, such as that prominently associated with foveal development in primates (14). Preliminary analysis of ganglion cell genotypes in whole-mounted chimeric retinas labeled with DNA probes (unpublished results) has yielded a more complete picture of this important cell class.
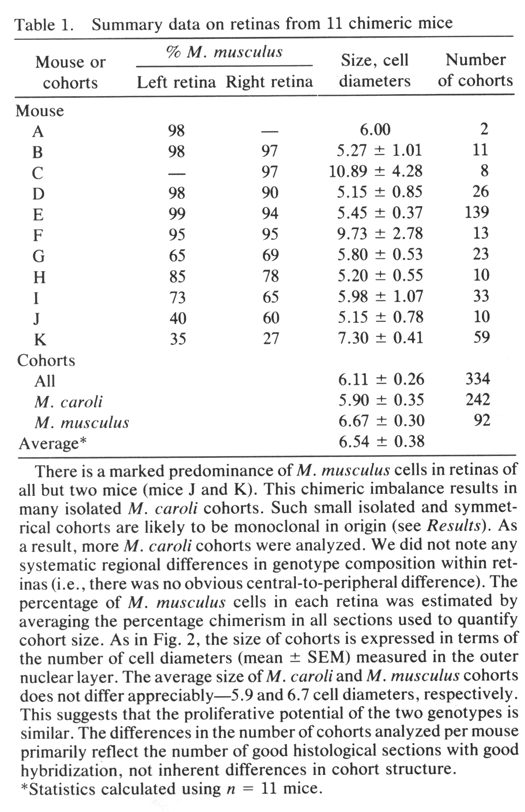
Chimeric Imbalance. Cells derived from M. musculus pre dominate in the retinas of 9 of 11 chimeras (Table 1) and on average comprise 80% of the total population of °ree;°ree;3.5 million cells that we estimate are in the adult mouse retina. Retinas from right and left eyes of chimeras have similar genotype ratios (Table 1). This finding provides support for the hy pothesis that right and left retinas originate from a single zone straddling the midline of the neural plate (15). M. musculus cells also predominate in ventral diencephalon (10); this suggests that the two tissues share a common origin in the neural plate. In several cases, the M. caroli genotype made up only 2—4% of the entire retina (Table 1). It is conceivable that in these highly imbalanced cases only a single M. caroli pro genitor contributed to the formation of the retina. However, even in these highly imbalanced chimeras, the minority genotype is widely distributed across the retina. This sug gests that cells of the two genotypes are mixed quite thor oughly during early stages of retinal development—probably between embryonic day (E) 9 and Eli, when the optic vesicles are budding out. The degree of mixing must be constrained at later stages, accounting for the discrete radi ally aligned units of M. musculus and M. caroli cells we see at maturity. The retention of sharp boundaries and vertical alignment are particularly impressive given the significant degree of retinal expansion that occurs after these cohorts are generated.
Clonal and Polyclonal Cohorts. Cohorts in all
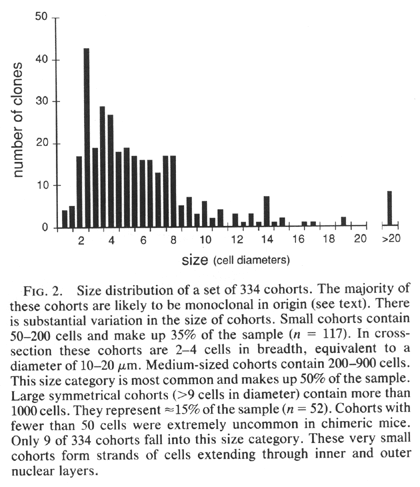
retinas vary greatly in size (Figs. 1 and 2). Each cohort originates either
from a single progenitor cell (a coherent clone) or from several progenitor
cells (a polyclone). We find a sharp cutoff in the minimum size of cohorts.
With very few exceptions (Fig. 2), the smallest cohorts in each retina are
2—4 cells wide and contain 100—200 cells (Figs. iF and 2). These cohorts
represent single ontogenetic quanta and are almost certainly derived from
single progenitors. These monoclonal cohorts were often clustered near each
other (Fig. iB, left side), as if two or more progenitors, descended from a
common progenitor, had moved apart a small distance. Large cohorts often
represent aggregates of ontogenetic units that share a common border at
maturity but do not share a single pro genitor. Because of the way in which
these polyclonal cohorts are combined by apposition of columns of progeny
cells, they will often lack symmetry in the retinal plane. As one would
predict, such cohorts are very common in bal anced chimeras, in which each
genotype contributes roughly half of the cells. However, many other large
cohorts—for example, the large cohort in Fig. iD (Table 1, mouse C)—may be
monoclonal in origin. This is particularly true in highly imbalanced
chimeras in which polyclonal aggregates of the minority genotype are
unlikely to form because the minority progenitors are diluted among a large
pool of the majority genotype (16, 17). In the imbalanced cases, we have
encountered isolated cohorts as large as 10—30 cells wide. They may be the
descendants of single progenitors whose progeny stayed together from early
stages of development. DISCUSSION
A Retinal Module. Both clones and polyclones in chimeric mice extend across all layers of the retina. The smallest cohorts, such as those illustrated in Fig. iB, appear to be perfectly aligned retinal core samples with sharply defined borders in both cell-rich layers. Each of these cohorts is a radial unit—in essence, a retinal building block (18). Their dimensional uniformity through the retinal layers indicates that the ratio of retinal cell types is also comparatively uniform. Polyclonal cohorts are simply combinations of these units, and they also appear to contain all cell types in approximately the same proportion as the retina itself. The only exception to this rule concerns rare retinal cell types. For example, horizontal cells make up only 1 in 300 retinal cells. Consequently, the smallest retinal clones may often lack horizontal cell representation. If a founder cell is to produce a complete retinal array it must give rise to at least 300 daughter cells. Number of Retinal Progenitors. Two questions arise: How large can a retinal clone get? How many founder cells contribute to the formation of the retina? The minimum size of the pool of proliferative cells that gives rise to the retina can be estimated using data on genotype ratios among our set of chimeras (19). In three retinas the M. musculus cells outnumber M. caroli cells by more than 25 to 1. This suggests that no less than 25 progenitor cells must be set aside from the pool of central nervous system progenitors to generate the retinas. The retina of the mouse is, therefore, probably made up of no less than 25 founder clones. Since the adult murine retina contains 3.0—3.5 million cells, each of these founder cells could generate clones containing as many as 140,000 cells. This is more than an order of magnitude larger than the largest coherent clones we have identified in the chimeric retina (Fig. 2). One explanation is that many more than 25 cells originally contribute to formation of retina. Another possibility is that this discrepancy is due to extensive mixing of cells of the two genotypes during early development. This mixing may split apart the progeny of the founder cells. When two sister cells become separated, they will give rise to two smaller coherent clones that are themselves related by de scent from the common founder. From this perspective, the size of a coherent clone may provide an index of its age or its hierarchy (see Figs. lB and 2). Clonal Uniformity. Small coherent clones in chimeric retina typically contain 100—200 cells and are solid radial arrays. In contrast, clones marked by Turner et al. (6) using a retroviral sequence incorporated by retinal progenitors on E13 are only rarely complete arrays of retinal cells and are not repre sentative of the retina. This is illustrated most clearly by the fact that none of 93 clones marked at E13 by retroviral transfection contains representatives of each of the six most common retinal cell types. One interpretation of this difference in clonal structure is that over a period of one or two cell divisions, probably at some time between Eli and E13, the majority of progenitors lose their ability to produce complete arrays of retinal cell types—that is, progenitor potential becomes restricted and, consequently, the types of cells progenitors produce become highly variable. Coherent clones in chimeras are presumably established at a time before lineage restriction. This would contribute to the marked difference in cellular composition of chimeric and retroviral cohorts. However, it is also possible that the difference in structure of chimeric and retroviral cohorts results from methodolog ical differences. The use of retroviral markers may yield experimental artifacts. For example, it has been shown that the expression of retroviral constructs can be suppressed by host cells (20). Selective suppression by different cell types could result in incomplete labeling of retinal arrays. Turner et al. (6) show a 6-fold difference in percentages of Muller glia labeled by transfection at birth with the HIS and ACT vectors. Furthermore, it is possible that insertion of retroviral sequences (21) or, for that matter, the intracellular injection of dyes (22), perturb subsequent cell proliferation and differ entiation. In this regard it is of interest to note that the average ratio of cells in inner to outer nuclear layers of the largest 10 clones marked in embryonic mouse by Turner et al. (6) is ito 17. These large clones are those one would predict to have a composition most like the retina itself. However, taking into account the sequence of cell production after E13, the predicted inner to outer cell ratio is °ree;°ree;1 (04(23). Contrary to expectation, the smallest retroviral clones, not the largest, more nearly approximate the normal inner to outer ratio. Microenvironmental Modulation of Retinal Cell Determination. The marked variability among labeled clones has led previous workers to argue that a cell's phenotype in the retina is determined by its microenvironment and that lineage has minimal, if any, influence on cell phenotype (3—6). The uni formity of clones and cohorts in chimeric retina suggests that the microenvironment may actually be comparatively uniform or evenly graded across the surface of the mouse retina. Alternatively, it is conceivable that retinal microenvironment is not uniform but that intrinsic lineage restriction nonetheless generates uniform retinal arrays. As is true in a number of other systems (24—26), this process of lineage restriction may, of course, be modulated by cellular interactions.
We thank Richard Cushing for expert technical assistance. This work was supported by the National Eye Institute.
Acknowledgements: 1. Ramon y Cajal, S. (1892) The Vertebrate Retina in The Verte brate Retina, Principles of Structure and Function, ed. Rodieck, R. W. (pub. 1973, Freeman, San Francisco), pp. 775—904. 2. Froriep, A. (1906) in Handbuch der Vergleichenden und Experimentellen Entwicklungslehre der Wirbeltiere, ed. Hertwig, 0. (Fischer, Jena, F.R.G.), pp. 139—261. 3. Turner, D. L. & Cepko, C. L. (1987) Nature (London) 328,131—136. 4. Wetts, R. & Fraser, S. E. (1988) Science 239, 1142—1145. 5. Holt, C. F., Bertsch, T. W., Ellis, H M & Hams, W A (1988) Neuron 1, 15—26. 6. Turner, D. L., Snyder, E. Y. & Cepko, C. L. (1990) Neuron 4,833—845. 7. Wassle, H. & Riemann, H. J. (1978) Proc. R. Soc. London Ser. B 200, 441—461. 8. Wikler, K. C. & Rakic, P. (1991) Nature (London) 351, 397—400. 9. Rossant, J., Vijh, K. M., Siracusa, L. D. & Chapman, V. M. (1983) J. Embryol. Exp. Morph ol. 73, 179—191. 10. Goldowitz, D. (1989) Neuron 3, 705—713. 11. Goldowitz, D. & Mullen, R. J. (1982) J. Neurosci. 2, 1474— 1485. 12. Siracusa, L. D., Chapman, V. M., Bennett, K. L. & Hastie, N. D. (1983) J. Embryol. Exp. Morphol. 73, 163—178. 13. Watanabe, T. & Raff, M. C. (1990) Nature (London) 332, 834—837. 14. Yuodelis, C. & Hendrickson, A. (1986) Vision Res. 26, 847—856. 15. Sanyal, S. & Zeilmaker, G. H. (1977) Nature (London) 265, 731—733. 16. Schmidt,G. H., Wilkinson, M. M.&Ponder,B. A. J.(1986)J. Embryol. Exp. Morphol. 91, 197—208. 17. West, J. (1975) J. Theor. Biol. 50, 153—160. 18. Rakic, P. (1988) Science 241, 170—176. 19. Rossant, J. (1990) Neuron 2, 323—334. 20. Jahner, 0., Stuhlmann, H., Stewart, C. L., Harbers, K., LUhIer, J., Simon, I. & Jaenisch, R. (1982) Nature (London) 298, 623—628. 21. Covarrubias, L., Nishida, Y., Terao, M., DEustachio, P. & Mintz, B. (1987) Mol. Cell. Biol. 7, 2243—2247. 22. Miller, 0. 5., Lau, Y.-T. & Horowitz, S. B. (1984) Proc. Natl. Acad. Sci. USA 81, 1426—1430. 23. Young, R. W. (1985) Anat. Rec. 212, 199—205. 24. Doe, C. Q. & Goodman, C. J. (1985) Dev. Biol. 111, 206—219. 25. Stent, G. S. & Weisblat, D. A. (1985) Annu. Rev. Neurosci. 8, 45—70. 26. Watanabe, T. & Raff, M. C. (1990) Neuron
2, 461—467.
Since 16 June 99
|
Neurogenetics at University of Tennessee Health Science Center
| Print Friendly | Top of Page |
Mouse Brain Library | Related Sites | Complextrait.org
