 |
| ||||||
|
| ||||||||
|
| ||||||||
Home  Publications Publications |
|
|
Note to the Reader HTML edition: Copyright © 1998 Robert W. Williams and Karl Herrup Originally published in The Annual Review of Neuroscience 11:423–453 (1988). Last revised Sept 28, 2001.
Print Friendly Introduction How an animal senses, perceives, and acts depends on the organization
and number of elements that make up its nervous system. Of the several
kinds of neural elements, ranging in size from ion channels to
cytoarchitectonic divisions, the neuron is the fundamental building block.
Understanding processes that control numbers of neurons in the brains of
different animals at different stages of development is therefore of great
importance. Two approaches can be taken to the problem of neuron number.
The first is a cellular and molecular approach that focuses on final
effects and final causes that regulate this variable in single species.
The second approach has a broader focus on evolutionary, ecological, and
bioenergetic reasons for, and consequences of, different strategies used
to control the size of neuron populations in different species. Here the
aim is to understand the diversity of strategies used to modify neuron
number in response to natural selection. This review is divided into three
sections. The first section provides an analysis of neuron number in
adults of different species. The second and third sections examine the two
principal processes that control neuron number during development—neuron
production and neuron elimination. The total number of neurons in the central nervous system ranges from
under 300 for small free-living metazoans such as rotifers and nematodes
(e.g., Martini 1912), 30–100 million for the common octopus and small
mammals such as shrews and mice (Young 1971, Campbell & Ryzen 1953,
Williams
2000), to well over 200 billion for whales and elephants. Estimates
for the human brain range between 10 billion and 1 trillion. The
imprecision in these estimates is due almost entirely to uncertainty about
the number of granule cells in the cerebellum, a problem that can be
traced back to a study by Braitenberg & Atwood (1958). More recent work by
Lange (1975) makes a reasonably accurate estimate possible: The average
human brain (1350 gm) contains about 85 billion neurons. Of these, 12 to
15 billion are telencephalic neurons (Shariff 1953), 70 billion are
cerebellar granule cells (Lange 1975), and fewer than 1 billion are
brainstem and spinal neurons. [A revision: In a beautiful quantitative
analysis of human cortex using the optical disector, Pakkenberg and
Gundersen (1997) have shown that the number of neocortical neurons ranges
from 15 to 31 billion and averages about 21 billion. Other forebrain
structures—primarily the hippocampal region, basal ganglia, and
thalamus—are likely to contain an additional 5–8 billion neurons. Total
neuron number in humans therefore probably averages 95–100 billion. What
is perhaps more remarkable is the normal two-fold difference in
neocortical neuron number among healthy adults of normal intelligence.]
Behavior complexity is not a function of body size. It follows that an
increase in body mass alone does not require a matched increase in numbers
of cells. Neurons could simply be larger and could branch more widely
(Tower 1954, Purves et al 1986). Nonetheless, larger individuals and
larger species generally do have larger brains that do contain more
neurons. Even though neurons are larger and packed more loosely in the
brains of large species (Holloway 1968, Lange 1975), the increase in brain
weight more than offsets the lower density. A 6000-g elephant brain has
two to three times as many neurons as does a 1350-g human brain. The number of neurons and their relative abundance in different parts
of the brain is a determinant of neural function and, consequently, of
behavior. Phyla whose members have larger brains and more neurons respond
to environmental change with a greater range and versatility of behavior (Jerison
1985). Orders of mammals with big brains, such as cetaceans and primates,
are more clever than those with little brains, such as insectivores and
marsupials. However, the correlation breaks down as we narrow the focus
and compare allied species and even individuals within species—the
exceptions obscure any trend. There is no generally valid equation that
relates neuron number to behavioral complexity. For instance, humans of
normal intelligence may have brains that weigh only half the average of
1350 gm, and in which there is no evidence for any compensatory increase
in neuron density, can have normal intelligence (Hechst 1932, Cobb 1965).
The nervous system of all vertebrates and many invertebrates is a
mosaic composed of hundreds to thousands of neuron subassemblies. To begin
systematic study of the cellular demography of the nervous system we need
reliable, objective methods to group neurons together. One way is to group
neurons by lineage. The clone of all labeled cells descended from a single
precursor cell, whatever the mixture of types, defines the group (Jacobson
& Hirose 1978, Weisblat et al 1978, Jacobson & Moody 1984, Sanes et al
1986, Turner & Cepko 1987). The strength of this method in terms of
examining relations between lines of neuronal descent, pattern of cell
divisions, and the final fate of neurons will be considered in the section
on neuron proliferation. Variation in the numbers of neurons in a nucleus or ganglion of a group
of animals belonging to the same species is often regarded as an
experimental nuisance, and in many cases it is not possible to separate
genuine intraspecific diversity from technical artifact (Konigsmark 1970,
Williams & Rakic, 1986, 1988 a,b). Unfortunately, this problem masks a
fundamental issue. Not only must the absolute number of neurons be
regulated during development, but within a population of interbreeding
individuals (a deme) there must also be mechanisms that ensure the
production and maintenance of adequate variation in numbers of neurons.
Without this variation there would be no evolutionary change in either
total or relative numbers of neurons (DeBrul 1960, Armstrong 1982). It is
important to appreciate that mechanisms that control neuron number also
control the range of variation and, ultimately, rates of brain evolution (Mayr
1963). Thus it is not surprising that whenever and wherever it has been
looked for with any persistence, natural variation in the numbers of
neurons has been found, even in the “invariant” nervous systems of
arthropods and annelids (e.g., Lubbock 1858, Hertweck 1931, Goodman 1979,
Macagno 1980). It is well accepted that the ratio of different types of neurons in
single nuclei and the ratio of neurons in interconnected parts of the
nervous system are key determinants of neuronal performance (Wimer et al
1976, Katz & Grenander 1982). However, the limits around which ratios may
vary without loss of function are in some cases substantial. For instance,
in primates, ratios of neurons in the richly interconnected
cytoarchitectonic zones of the occipital lobe (visual areas 17, 18, and
19) vary by more than a factor of two (van Essen et al 1984, 1986,
Williams & Rakic 1986), and an equal magnitude of variation is found in
ratios of neurons in the peripheral nervous system (Ebbesson 1965). Great
variation is also found between ratios of homologous populations of
neurons in different species. Here the differences have an easily
recognized basis; brains are customized for the body and behavior of each
species and consequently ratios of cells will often be radically
different. For example, the ratio between granule cells and Purkinje cells
rises from less than 200 to 1 in mice to 3000 to 1 in humans (Wetts &
Herrup 1982, Lange 1975). A variety of studies have shown that a surplus of neurons does not give
rise to maladaptive behavior (e.g., Hollyday & Hamburger 1976, Chalupa et
al 1984, Ellis & Horvitz 1986); on the contrary, any supernumerary neurons
or glial cells can be sequestered or integrated into existing neuronal
circuitry and may easily result in more adaptive behaviors. This is as
true for roundworms (Ellis & Horvitz 1986) as for rats (Zamenhof 1942). A
20% surplus of neurons in Caenorhabditis elegans caused by a
mutation that blocks cell death results in no behavioral deficits in
moving, mating, or egg laying (Ellis & Horvitz 1986). What then are the
selective pressures that set an upper limit? In most tissues a deficit of a particular cell type can be corrected by
generating new cells, by coaxing cells into the depleted region from
surrounding tissues, or by modifing existing cells. As a rule, this is not
the case in the nervous system. Precursor cells capable of producing
neurons are either few in number or simply do not exist at late stages of
development (Miale & Sidman 1961, Rakic 1985). And neurons themselves
cannot divide—they are postmitotic cells unable to reenter a cycle of cell
division. Consequently, the number of neurons within each part of the
nervous system is, with some interesting exceptions, determined at early
stages of development, often without the benefit of any exposure to the
environment—an odd situation for a organ system designed to deal with the
environment. Mature neurons have only limited capabilities to change type
or move around. Thus, in contrast to cells in most other tissues, the
number, type, and distribution of neurons cannot be regulated about some
optimum at maturity. The production and deployment of neurons has to be
done right the first time, not an easy job given the extraordinary
complexity of the nervous system. Intrinsic FactorsIn leeches and nematodes, neuron number is under tight control of an
inflexible pattern of cell lineage. Shankland and Weisblat (1984) have
shown in the leech that if one of a bilateral pair of precursor cells (teloblasts)
is eliminated, descendents of the contralateral homologue will
occasionally cross the midline. However, this action depletes the donor
side to the benefit of the operated side. Up-regulation of cell number
either by proliferation or recruitment evidently is not possible, and
consequently the question of control of neuron number is rarely raised
explicitly in this work. In additional studies the stereotypic patterns of
cell lineage were perturbed either by mutation (Horvitz and Sulston, 1980)
or by the deletion of cells (Sulston & Horvitz 1977, Kimble & White 1980,
Weisblat & Blair 1982). These manipulations have revealed that, while for
the great majority of cells lineage constrains their developmental
potential to a single fate, for a few other neuroblasts cell-cell
interactions specify which of two or three alternate fates—frequently
arranged hierarchically—is expressed. In these experiments, the
manipulations alter the morphological and numerical fate of the lineage
lower on the hierarchy. Thus, again, their is no up-regulation of cell
number. Single gene effects. Given the relatively large size and
complexity of most higher vertebrates and their seemingly non-determinate
plan of development, they would seem to be an unlikely place to begin a
search for evidence of intrinsic cellular mechanisms controlling rates and
fractions of mitotic cells. Yet some of the strongest evidence for
intrinsic control of this type come from work on mammals; in particular,
from the mouse, and from genetic analyses of the development of this
species. A powerful tool in this approach has been the use of genetic
mosaics, specifically, the aggregation chimera. This experimental animal
is formed by bringing two embryos together at the 8-cell stage of
development, coaxing them to form a single embryo and then transplanting
the double-blastocyst to a recipient mother where it adjusts in size and
finishes gestation.
(PC in the chimera – PC in sg/sg)
Lineage-related control of neuron number. These conclusions lead
to the notion that specific lineages require specific genes for the
regulation of cell number. This raises the question of whether ensembles
of lineage-specific factors are involved in the regulation of neuron
number. Using lurcher (see above) as a cell marker, Herrup and Wetts
(1982c) counted the number of Purkinje cells in a series of lurcher<->wild-type
chimeras. Since the cell-autonomous action of the lurcher gene destroys
all of the lurcher Purkinje cells, the only cells they could count would
have descended from the non-lurcher (wild-type) embryo. They discovered
that the counts fell on integral multiples of the value of 10,200 and
suggested that this value represented numerical evidence that the
population of Purkinje cells consisted of a small number of clones of
cells. The number of cells in any one clone is the same and the apparent
quantal nature of the Purkinje cells of one genotype in these chimeras
reflects the selection of increasing numbers of wild-type cells to found
the clones. Extrinsic FactorsThe evidence cited above suggests that intrinsic, lineage autonomous
regulation of neuron number occurs during normal development. This
evidence however, does not preclude a role for extrinsic factors. There
are, however, few factors that have been identified as either mitogens or
mitotic inhibitors that might serve as humoral regulators of neuronal cell
division. Nerve growth factor was once thought to be a mitogen of neurons
in the peripheral nervous system, but is now known to exert its effects
primarily through prevention of cell death and concomitant hypertrophy of
the cells (Thoenen & Barde 1980, Thoenen et al 1985). Growth hormone and
sex hormones are both candidates for substances with neuronal mitogenic
effects. Perhaps the best studied compound of this class is the thyroid
hormone, thyroxine. Control of Neuron Number by Cell DeathOne of the most counterintuitive and seemingly wasteful processes in brain development is the death of an often large fraction of the initial complement of neurons and glial cells. First noted by Studnicka (1905), Collin (1906), and von Szily (1912) and others in the vertebrate central nervous system, serious attention to this process and the proof that many dying cells are young neurons was delayed until catalytic studies by Glücksmann (1940), Romanes (1946), Hamburger and Levi-Montalcini (1946), Beaudoin 1955, and Hughes (1961). The literature has been particularly well reviewed (Källén 1965, Hughes 1968; Prestige 1970, Cowan 1973, Silver 1978, Jacobson 1978, Cunningham 1982, Berg 1982, Beaulaton & Lockshin 1983, Hamburger & Oppenheim 1983, Horvitz et al 1982, Truman 1984, Oppenheim 1987).
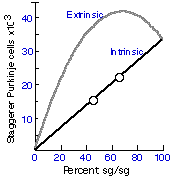
VariationNeuron death is by no means universal throughout the animal kingdom.
The process in unknown in Aplysia californica (Jacob 1984), and is
also rare or unknown in elasmobranchs (sharks and rays), teleosts, or
reptiles (but see Fox & Richardson 1982). There is no evidence of motor
neuron loss in zebrafish, dogfish, or stingray (reviewed in Mos &
Williamson 1986). With the important exception of birds, mammals, and
nematodes, it appears that the elimination of neurons is most pronounced
in species that undergo metamorphosis, and is least pronounced in
vertebrates and invertebrates that do not metamorphose and that grow
throughout life. Comparative Perspective on Roles of Neuron DeathIn nematodes neuron death is not contingent on the status of other
cells. Death is an inflexible autonomous fate that serves to eliminate
neurons that are apparently unwanted byproducts of patterns of cell
lineage. In C. elegans one common mechanism, involving the action
of at least four ced gene products, catalyzes the death of most
cells—neurons included. Mutations of the genes ced-3 and ced-4
prevent degeneration and death entirely (Hedgecock et al 1983, Ellis &
Horvitz 1986). The most probable reason why neurons die in nematodes is
not to remove potentially detrimental neurons, but to rid the organisms of
unneeded, metabolically demanding cells. In this animal, neuron
elimination appears to be strictly a means to regulate number, not to
regulate ratios of cells or to improve neuronal performance. However, in
many, if not most other phyla, the process of cell elimination in any one
population of neurons is regulated by several different kinds of
interactions, each designed to optimize cell number or cell performance
along a different parameter. Here, neuron death is just the final common
pathway of disparate processes having disparate causes. The relative
contributions of these processes will differ from animal to animal, from
neuron population to neuron population. Neuron Death and Neuron RatiosThe rate and duration of neuron production in distant populations that
are ultimately interconnected may not be well matched. This will produce
quantitative imbalances.4
Cell elimination provides a way to adjust ratios of cells after a link
between interconnected populations by axons or dendrites has been
established. The larger the nervous system and the more disjoint the
production of interconnected cell populations (e.g., motor neurons and
muscle fibers, pre- and post-ganglion sympathetic neurons, receptor
neurons and central target cells), the more important secondary
fine-tuning will be. For example, motor neurons in vertebrates are lost
shortly after their axons have reached target musculature. If the target
is removed early enough in development (before or during the period of
cell death) then the severity of neuron loss is increased. Cell death can
wipe out the entire population if the target is removed early enough in
development (e.g. Hughes & Tschumi 1954, Lancer & Fallon 1984).
Complementary studies in which the the size and number of target cells is
increased have been carried out in several systems (e.g., Hamburger 1939,
Hollyday and Hamburger 1976, Pilar et al 1980, Boydston and Sohal 1979,
Narayanan and Narayanan 1978, Lamb 1979, Chalupa et al. 1984, Maheras &
Pollack 1985). In all cases, an increase in number of targets cells causes
an appreciable increase in the percentage of surviving neurons, but the
increase is never as great as would be required to maintain a normal
ratios between interconnected cells. Thus, neuron elimination even in
these systems is not regulated entirely by extrinsic factors. Some
fraction of the loss is evidently intrinsic and thus cannot be regulated
during ontogeny to bring about the most adaptive ratio between cells.
However, the long term selective pressures on a species may result in
rather rapid change in the importance of intrinsic control. As pointed out
by Prestige (1970), the relative weight of intrinsic and extrinsic control
varies substantially between vertebrate classes. For example, the loss of
thoracic motor neurons in the anuran spinal cord is heavily dependent on
the status of the periphery, whereas the loss of the same population of
neurons in chick and mouse appears to be largely under intrinsic control.
This specific difference may be related to the fact that anurans
metamorphose, whereas birds and mammals develop directly. SummaryIn comparing strategies used to control neuron number, we find it useful to view nervous system development as occurring in three phases. The phases overlap—each is a process, not an event. The first phase is the development of a genetic nervous system. This is a nervous system of simple genetic intention; not a blueprint or pile of bricks. Its characteristics are abstract: how many neurons is this stem cell destined to produce, which cells are programmed to die, how much target does this cell need to survive. At the level of the individual, the genetic nervous system is essentially fixed, but over generations it is fluid. As this genetic intent interacts within a world of cells, a real brain to appears. A new set of rules only tacitly present in the genome is expressed—the embryonic nervous system emerges. The interaction of its parts defines the shape and size of the nervous system. Neuron numbers are adjusted interactively by changes in proliferative potential and the severity of cell death. Cell fates are established in part through interactions with other cells and with hormones. Glial cell numbers are adjusted to match the neuron populations. This phase of brain development is what the embryologist sees under the microscope. The final phase of development begins when the brain starts to function and the animal starts to deal with its world. Small changes in neuron number may occur.during this period, but the changes are generally of minor functional importance. At this point, the smaller elements of neuronal organization are refined in shape, number, and distribution. Axons are lost or rearranged; dendrites grow, branch, and retract; synapses are fine-tuned; and finally, receptors densities and transmitter titres are adjusted. Some of these interactions and numerical adjustments continue until the animal dies.
ReferencesAllen, C. Sievers, J., Berry, M., Jenner, S. 1981. Experimental studies on cerebellar foliation. II. A morphometric analysis of cerebellar fissuration defects and growth retardation after neonatal treatment with 6-OHDA in the rat. J. Comp. Neurol. 203: 771–83 Ambros, V., Horvitz, H.R. 1984 Heterochronic mutants of the nematode Caenorhabditis elegans. Science 226: 409–16 Anderson, H., Edwards, J.S., Palka, J. 1980. Developmental neurobiology of invertebrates. Ann. Rev. Neurosci. 3: 97–139 Angelo, J.C., Prothero, J.W. 1985. Clonal attenuation in chick embryo fibroblasts. Experimental data, a model and computer simulations. Cell Tissue Kinet. 18: 27–43 Armstrong, E. 1982. Mosaic evolution in the primate brain: Differences and similarities in the hominoid thalamus. In Primate Brain Evolution. Methods and Concepts. ed. E. Armstrong, D. Falk, pp. 131–61. New York: Plenum Press Armstrong, E. 1983. Relative brain size and metabolism in mammals. Science 220: 1302–4 Armstrong, E., Bergeron, R. 1985. Relative brain size and metabolism in birds. Brain Behav. Evol. 26: 141–53 Armstrong, R.C., Clarke, P.G.H. 1979. Neuronal death and the development of the pontine nuclei and inferior olive in the chick. Neuroscience 4: 1635–47 Ashwell, K.W. and Watson, C .R.R. (1983). The development of facial motoneurones in the mouse—Neuronal death and the innervation of the facial muscles. J. Embryol. Exp. Morphol. 77: 117–41 Beach, D.H., Jacobson, M. 1979. Influence of thyroxine on cell proliferation in the retina of the clawed frog at different ages. J. Comp. Neurol. 183: 615–24 Beaudoin, A.R. 1955. The development of lateral motor column cells in the lumbo–sacral cord in Rana pipiens, I. Normal development and development following unilateral limb ablation. Anat. Rec. 121: 81–95 Beaulaton, J., Lockshin, R.A. 1982. The relation of programmed cell death to development and reproduction: Comparative studies and an attempt at classification. Int. Rev. Cytol. 79: 215–35 Beddington, R.S.P. 1981. An autoradioagraphic analysis of the potency of embryonic ectoderm in 8th day postimplantation mouse embryos. J. Embryol. Exp. Morph. 69: 265–85 Beddington, R.S.P. 1983. Histogenic and neoplastic potential of different regions of mouse embryonic egg cylinder. J. Embryol. Exp. Morph. 75: 189–204 Berg, D.K. (1982) Cell death in neuronal development. Regulation by trophic factors. In Neuronal Development, ed. N.C. Spitzer, pp. 297–331. New York: Plenum Blinkov, S.M., Glezer, I.I. 1968. The Human Brain in Figures and Tables: A Quantitative Handbook. New York: Basic Books, Inc. Plenum Press Block, J.B., Essman, W.B. 1965. Growth hormone administration during pregnancy: A behavioural difference in offspring rats. Nature 205:1136–37 Boydston, W.R., Sohal, G.S. 1979. Grafting of additional periphery reduces embryonic loss of neurons. Brain Res. 178: 403–10 Bradely, P., Berry, M. 1978 Purkinje cell dendritic tree in mutant mouse cerebellum. A quantitative Golgi study of weaver and staggerer mice. Brain Res. 142: 135–41. Braitenberg, V., Atwood, R.P. 1958. Morphological observations on the cerebellar cortex. J. Comp. Neurol. 109: 1–33. Breedlove, S.M., Arnold, A.P. 1980. Hormone accumulation in a sexually dimorphic motor nucleus of the rat spinal cord. Science 210: 564–566. Bullock, T.H., Horridge, G.A. 1965 Structure and Function in the Nervous System of Invertebrates. San Francisco: Freeman. Bush G.L., Case S.M., Wilson, A.C., and Patton J.L 1977. Rapid speciation and chromosomal evolution in mammals. Proc. Natl. Acad. Sci. USA 74: 3942–46 Caddy, K.W.T.& Biscoe,T.J. 1979. Structural and quantitative studies on the normal C3H and Lurcher mutant mouse. Phil T. Roy. Soc. Lond Ser. B 287: 167–201 Campbell, B., Ryzen, M. 1953. The nuclear anatomy of the diencephalon of Sorex cinereus. J. Comp. Neurol. 99: 1–22 Chalfie, M., Horvitz, H.R., Sulston, J.E. 1981. Mutations that lead to reiterations in the cell lineage of C. elegans. Cell 24: 59–81 Chalupa, L.M., Williams, R. W., Henderson, Z. 1984. Binocular interaction in the fetal cat regulates the size of the ganglion cell population. Neuroscience 12: 1139–46 Champy, C. 1922. L'action de l'extrait thyroidien sur la multiplication cellulaire. Caracter electif de cette action. Arch. Morphol. Gen. Exper. 4: 1–58 Chan, W.Y., Tam, P.P.L. 1986. The histogenetic potential of neural plate cells of early somite–stage mouse embryos. J. Embryol. Exp. Morphol. 96: 183–93 Chiarodo, A.T. 1963. The effects of mesothoracic leg disc extirpation on the postembryonic development of the nervous system of the blowfly Sarcophaga bullata. J. Exp. Zool. 153: 263–77 Clarke, P.G.H., Cowan W.M. 1976. The development of the isthmo–optic tract in the chick, with special reference to the occurrence and correction of developmental errors in the location and connection of isthmo–optic neurons. J. Comp. Neurol. 167:143–64 Clendinnen, B.G., Eayrs, J.T. 1961. The anatomical and physiological effects of prenatally administered somatotrophin on cerebral development. J. Endocrinol. 22:183–93 Cline, H.T., Constantine–Paton, M. 1986. Thyroxine effects on the development of the retinotectal projection. Soc. Neurosci. Abs. 12:437 Cobb, S. 1965. Brain size. Arch. Neurol. 12:555–61 Collin, R. 1906–1907. Recherches cytologiques sur le developpement de la cellule nerveus. Nevraxe 8:181–303 Cooke, J. 1980. Early organization of the central nervous system: Form and pattern. Curr. Top. Devel. Biol. 15:373–407 Cowan, W.M. 1973. Neuronal death as a regulative mechanism in the control of cell number in the nervous system. In Development and Aging in the Nervous System, ed. M. Rockstein, pp. 19–41. New York: Academic Press Crespo, D., O'Leary, D.D.M., Cowan, W.M.1985. Changes in the number of optic nerve fibers during late prenatal and postnatal development in the albino rat. Dev. Brain Res. 19:129–34 Cunningham, T.J. 1982. Naturally occurring neuron death and its regulation by developing neural pathways. Internat. Rev. Cytol. 74: 163–86. Das, G.D. and Pfaffenroth, M.J. 1977. Experimental studies on the postnatal development of the brain. III. Cerebellar development following localized administration of ENU. Neuropath. Appl. Neurobiol. 3:191–212 DeBrul, L. 1960. Structural evidence in the brain for a theory of the evolution of behavior. Perspect. Biol. Med. 4:40–57. Delhaye–Bouchaud, N., Geoffroy, B., Mariani, J. 1985. Neuronal death and synapse elimination in the olivocerebellar system. I. Cell counts in the inferior olive of developing rats. J. Comp. Neurol. 232:299–308 DeLong, G.R., Sidman, R.L. 1962 Effects of eye removal at birth on histogenesis of the mouse superior colliculus: An autoradiographic analysis with tritiated thymidine. J. Comp. Neurol. 118:205–23 Diglio, T., Herrup, K. 1982. A significant fraction of the adult number of mature cerebellar Purkinje cells first appears between postnatal days 16 and 30 in the mouse. Soc. Neurosci. Abst. 8:636 Dobbing, J., Smart J.L. 1974. Vulnerability of developing brain and behaviour. Brit. Med Bull. 30:164–68. Easter, S.S., Jr., Rusoff, A.C., Kish, P.E. 1981. The growth and organization of the optic nerve and tract in juvenile and adult goldfish. J. Neurosci. 1:793–811. Ebbesson, S.O.E. 1965. Quantitative studies of superior cervical sympathetic ganglia in a variety of primates including man. I. The ratio of preganglionic fibers to ganglionic neurons. J. Morph. 124:117–32 Eisenberg, J.F. 1981. The Mammalian Radiations. An Analysis of Trends in Evolution, Adaptation, and Behavior. Chicago: The University of Chicago Press Eisenberg, J.F., Wilson D.E. 1982. Relative brain size and feeding strategies in the Chiroptera. Evolution 32: 740–51 Ellis, H.M., Horvitz, H.R. 1986. Genetic control of programmed cell death in the nematode C. elegans. Cell 44:817–29 Faber, D.S., Korn, H., eds. 1978. Neurobiology of the Mauthner Cells. New York: Raven Press Fankhauser, G., Vernon, J.A., Frank, W.H., Slack, W.V. 1955. Effect of size and number of brain cells on learning in larvae of the salamander, Triturus viridescens. Science. 122:692–93 Finlay, B.L., Slattery, M. 1983. Local differences in the amount of early cell death in neocortex predict adult local specializations. Science 219:1349–51 Finlay, B.L., Wikler, K.C., Sengelaub, D.R. 1987. Regressive events in brain development and scenarios for vertebrate brain evolution. Brain Behav. Evol. 30:102–17 Fischbach, K.F., Heisenberg, M. 1981. Structural brain mutant of Drosophila melanogaster with reduced cell numbers in the medulla cortex and with normal optomotor yaw response. Proc. Natl. Acad. Sci. USA 78:1105–9 Fischbach, K.F., Technau, G. 1984. Cell degeneration in the developing optic lobes of the sine oculis and small–optic–lobes mutants of Drosophila melanogaster. Devel. Biol. 104:219–39 Fox, G.Q., Richardson, G.P. 1982. The developmental morphology of Torpedo marmorata: Electric lobe–electromotoneuron proliferation and cell death. J. Comp. Neurol. 207:183–90 Gao, W–Q., Macagno, E. R. 1986. Extension and retraction of axonal projections by existence of neighboring homologues. I. The HA cells. J. Neurobiol. 18: 43–59 Glucksmann, A. 1940. Development and differentiation of the tadpole eye. Brit. J. Ophthal. 24:153–78 Glucksmann, A. 1951. Cell deaths in normal vertebrate ontogeny. Biol. Rev. 26:59–86 Goldschmidt, R. (1909) Das Nervensystem von Ascaris lumbridoides und megalodephala. II. Z. Wiss. Zool. 92:306–57 Goldman, S.A., Nottebohm F. 1983. Neuronal production, migration, and differentiation in a vocal control nucleus of the adult canary brain. Proc. Natl. Acad. Sci. USA 80:2390–94 Goodman, C. 1976. Constancy and uniqueness in a large population of small interneurons. Science 193:502–4 Goodman, C.S. 1977. Neuron duplications and deletions in locust clones and clutches. Science 197:1384–86 Goodman, C.S. 1978. Isogenic grasshoppers: Genetic variability in the morphology of identified neurons. J. Comp. Neurol. 182:681–706 Goodman, C.S. 1979 Isogenic grasshoppers: Genetic variability and development of identified neurons. In Neurogenetics: Genetic Approaches to the Nervous System, ed. X.O. Breakfiled, pp. 101–51. New York: Elsevier–North Holland Gould S.J. 1977. Ontogeny and Phylogeny. The Belknap Press of Harvard University Press, Cambridge, Mass Graziadei, P.P.C., Monti Graziadei, G.A. 1979 a. Neurogenesis and neuron regeneration in the olfactory system of mammals. I. Morphological aspects of differentiation and structural organization of the olfactory sensory neurons. J. Neurocytol. 8:1–18 Graziadei, P.P.C., Monti Graziadei, G.A. 1979b. Neurogenesis and neuron regeneration in the olfactory system of mammals. II. Degeneration and reconstitution of the olfactory sensory neurons after axotomy. J. Neurocytol. 8:197–213 Guesella, J., Geller, R., Clarke, B., Weeks, V., Housman, D. 1976. Commitment to erythroid differentiation by Friend erythroleukemia cells. A stochastic analysis. Cell 9:221–29 Hamburger, V. 1939. Motor and sensory hyper–plasia following limbbud transplantations in chick embryos. Physiol. Zool. 12:268–284 Hamburger, V. 1975. Cell death in the development of the lateral motor column of the chick embryo. J. Comp. Neurol. 160:535–46 Hamburger, V., and Levi–Montalcini, R. 1949. Proliferation, differentiation and degeneration in the spinal ganglia of the chick embryo under normal and experimental conditions. J. Exp. Zool. 111: 457–501 Hamburger, V., Oppenheim, R. W. 1982. Naturally occurring neuronal death in vertebrates. Neurosci. Comm.1: 39–55 Hechst, B. 1932. Uber einen Fall von Mikroencephalie ohne geistigen Defekt. Arch. Psychiat. u. Nervenk. 97:64–76 Hedgecock, E., Sulston, J.E., Thompson, N. 1983. Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science 220:1277–80 Herrup, K. 1986. Cell lineage relationships in the development of the mammalian CNS: Role of cell lineage in control of cerebellar Purkinje cell number. Devel. Biol. 115:148–54 Herrup, K. 1983. Role of staggerer gene in determining cell number in cerebellar cortex. I. Granule cell death is an indirect consequence of staggerer gene action. Devel. Brain Res. 11:267–74 Herrup, K., 1987 Roles of cell lineage in the developing mammalian brain. Curr. Top. Devel. Biol 21: 65–97 Herrup, K., Mullen, R.J. 1979. Staggerer chimeras: Intrinsic nature of Purkinje cell defects and implications for normal cerebellar development. Brain Res. 178:443–57 Herrup, K., Mullen, R.J. 1981. Role of the staggerer gene in determining Purkinje cell number in the cerebellar cortex of mouse chimeras. Dev. Brain Res. 1:475–85 Herrup, K., Wetts, R., Diglio, T.J. 1984a. Cell lineage relationships in the development of the mammalian CNS. II. Bilateral independence of CNS clones. J. Neurogenet. 1:275–88 Herrup, K., Letsou, A., Diglio, T.J. 1984b. Cell lineage relationships in the development of the mammalian CNS: The facial nerve nucleus. Devel. Biol. 103:329–36 Herrup, K., Sunter, K. 1986 Cell lineage dependent and independent control of Purkinje cell number in the mammalian CNS: Further quantitative studies of Lurcher chimeric mice. Devel. Biol 117: 417–27 Herrup, K , Sunter, K. 1987. Numerical matching during cerebellar development: Quantitative analysis of granule cell death in staggerer mouse chimeras. J. Neurosci. 7:829–836 Hertweck, H. 1931. Anatomie und Variabilitat des Nervensystems und der Sinnesorgane von Drosophila melanogaster (Meigen). Z. Wiss. Zool. 139:559–663 Hinds, J.W., Hinds P.L. 1978. Early development of amacrine cells in the mouse retina: An electron microscopic, serial section analysis. J. Comp. Neurol. 179: 277–300 Hofbauer, A., Campos–Ortega, J.A. 1976. Cell clones and pattern formation: Genetic eye mosaics in Drosophila melanogaster. Wilhelm Roux Arch. Devel. Biol. 179:275–89 Holloway, R.L. 1968. The evolution of the primate brain: Some aspects of quantitative relations. Brain Res. 7:121–72 Holloway, R.L. 1980. Within–species brain–body weight variability: A reexamination of the Danish data and other primate species. Am. J. Phys. Anthropol. 53:109–21 Hollyday, M., Hamburger, V. 1976. Reduction of the naturally occurring motor neuron loss by enlargement of the periphery. J. Comp. Neurol. 170:311–20 Horvitz, H.R., Ellis, H.M., Sternberg P.W. 1982. Programmed cell death in nematode development. Neurosci. Comment. 1:56–65 Hughes, A.F. 1957. The development of the primary sensory system in Xenopus laevis (Daudin). J. Anat. 91:323–38 Hughes, A.F. 1961. Cell degeneration in the larval ventral horn of Xenopus laevis. J. Embryol. Exp. Morph. 9:269–84 Hughes, A. 1968. Aspects of Neural Ontogeny. New York, Academic Press Hughes, A., Tschumi, P.A. 1958. The factors controlling the development of the dorsal root ganglia and ventral horn in Xenopus laevis (Daud.) J. Anat. 92:498–527 Jacob, M.H. (1984) Neurogenesis in Aplysia californica resembles nervous system formation in vertebrates. J. Neurosci. 4:1225–39 Jacobs, D.S., Perry, V.H., Hawken, M.J. (1984) The postnatal reduction of the uncrossed projection from the nasal retina in the cat. J. Neurosci. 4: 2425–33 Jacobson, M. 1978. Developmental Neurobiology. 2nd ed., New York: Plenum Press Jacobson, M. 1985. Clonal analysis and cell lineages of the vertebrate central nervous system. Ann. Rev. Neurosci. 8:71–102 Jacobson, M., Hirose, G. 1978. Origin of the retina from both sides of the embryonic brain: A contribution to the problem of crossing at the optic chiasma. Science 202:637–39 Jacobson, M., Moody, S.A. 1984. Quantitative lineage analysis of the frog's nervous system. I. Lineages of Rohon–Beard neurons and primary motoneurons. J. Neurosci. 4:1361–69 Jerison, H.J. 1955. Quantitative analysis of evolution of the brain in mammals. Science 133:1012–14 Jerison, H. J. 1973. Evolution of the Brain and Intelligence. New York. Academic Press Jerison, H.J. 1985. Animal intelligence as encephalization. Phil. Trans. R. Soc. Lond.Ser. B. 308:21–35 Källén, B. 1955 Cell degeneration during normal ontogenesis of the rabbit brain. J. Anat. 89:153–62 Källén, B. 1965. Degeneration and regeneration in the vertebrate central nervous system during embryogenesis. Prog. Brain Res. 14:77–96 Kaplan, M.S. 1981. Neurogenesis in the 3–month–old rat visual cortex. J. Comp. Neurol. 195:323–38 Katz, M.J., Lasek, R.J. 1978. Evolution of the nervous system: Role of ontogenetic mechanisms in the evolution of matching populations. Proc. Natl. Acad. Sci. USA 75: 1349–52 Katz, M.J., Grenander, U. 1982. Developmental matching and the numerical matching hypothesis for neuronal cell death. J. Theor. Biol. 98:501–17 Kety, S.S., Schmidt, C.F. 1948. The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J. Clin. Invest. 27:484–492 Kimmel, C.B., Eaton, R.C. 1976. Development of the Mauthner cell. IN Simpler Networks and Behavior. ed. J.C. Fentress, pp. 186–202. Sunderland, Mass, Sinauer Kolb, H., Nelson, R., Mariani, A. 1981. Amacrine cells, bipolar cells and ganglion cells of the cat retina: A Golgi study. Vision Res. 21:1081–1114 Kollros, J.J. 1953 The development of the optic lobes in the frog. I. The effects of unilateral enucleation in embyronic stages. J. Exp. Zool 123: 153–87 Kollros, J.J., McMurray, V.M. 1955. The mesencephalic V nucleus in anurans. J. Comp. Neurol. 102:47–61 Kollros, J.J. 1982. Peripheral control of midbrain mitotic activity in the frog. J. Comp. Neurol. 205:171–178 Kollros, J.J., Thiesse M.L. 1985. Growth and death of cells of the mesencephalic fifth nucleus in Xenopus laevis larvae. J. Comp. Neurol. 233:481–489 Konigsmark, B.W. 1970. The counting of neurons. In Contemporary Research Methods in Neuroanatomy, eds. W.J.H. Nauta and S.O. Ebbesson, pp. 315–40. New York: Springer Verlag Konishi, M. 1985. Birdsong: From behavior to neuron. Ann. Rev. Neurosci. 8:125–70 Krebs, H.A. 1950 Body size and tissue respiration. Biochim. Biophys. Acta 4:249–269 Kreisman, N.R., J. E. Olson, D.S. Horne, Holtzman, D. 1986. Developmental increases in oxygen delivery and extraction in immature rat cerebral cortex. Neurosci. Abs. 12:451. Lamb, A.H. 1979. Evidence that some developing limb motoneurons die for reasons other than peripheral competition. Dev. Biol. 71:8–21 Lande, R. 1979. Quantitative genetic analysis of multivariate evolution, applied to brain:body size allometry. Evolution 33:402–416 Landis, D.M.D. and Sidman, R.L. 1978. Electron microscopic analysis of postnatal histogenesis in the cerebellar cortex of staggerer mutant mice. J. Comp. Neurol. 179:831–863. Lange, W. 1975 Cell number and cell density in the cerebellar cortex of man and other mammals. Cell Tiss. Res. 157: 115–24 Lanser, M.E., Fallon, J.F.1984. Development of the lateral motor column in the limbless mutant chick embryo. J. Neurosci. 4:2043–50 Lanser, M.E., Fallon, J.F. 1987 Development of the branchial lateral notor column in the Wingless mutatnt chick embryo: Motoneuron survival under varuying degrees of peripheral load. J. Comp. Neuro. 261: 423–34 Lapicque, L. 1907. Le poids encephalique en fonction du poids corporel entre individus d'une meme espece. Bull. Mem. Soc. Anthropol. Paris. 8:313–45 Lauder, J.M., Altman, J., Krebs, H. 1974. Some mechanisms of cerebellar foliation: effects of early hypo– and hyperthyroidism. Brain Res. 76:33–40 Lauder, J.M., Krebs, H. 1976. Effects of p–chlorophenylalanine on time of neuronal origin during embryogenesis in the rat. Brain Res. 107:638–44 Lauder, J.M., Krebs, H. 1978. Serotonin as a differentiation signal in early neurogenesis. Dev. Neurosci. 1:15–30 Lemke, G.E., Brokes, J.P. 1983. Identification and purification of glial growth factor. J. Neurosci. 4:75–83 Levi–Montalcini, R. 1950. The origin and development of the visceral system in the spinal cord of the chick embryo. J. Morphol. 86:253–83 Linden, R., Perry, V.H. 1982. Ganglion cell death within the developing retina: A regulatory role for retinal dendrites? Neuroscience 7:2813–27 Lillegraven, J.A. 1979 Reproduction in Mesozoic mammals. In Mesozoic Mammals: The First Two–Thirds of the Mammalian History, eds. J.A. Lillegraven, Z. Kielan–Javorowska, W.A. Clemens, pp. 259–76. Berkeley, Calif. University of California Press Lubbock, J. 1858. Variability of the nervous system. Proc. R. Soc. Lond. 9:480–86 Macagno, E.R., Lopresti, V., Levinthal, C. 1973. Structure and development of neuronal connections in isogenic organisms: Variation and similarities in the optic system of Daphnia magna. Proc. Natl. Acad. Sci USA 70:57–61 Macagno, E.R. 1979. Cellular interactions and pattern formation in the development of the visual system of Daphnia magna (Crustacea, Branchiopoda). I. Interactions between embryonic retinular fibers and laminar neurons. Dev. Biol. 73:206–38 Macagno, E.R. 1980. Number and distribution of neurons in leech segmental ganglia. J. Comp. Neurol. 190:283–302 Maheras, H.M., Pollack, E.D. 1985. Quantitative compensation by lateral motor column neurons in response to four functional hindlimbs in a frog tadpole. Dev. Brain Res. 19:150–54 Mallet, J., Huchet, M., Pougeois, R., and Changeux, J.P. 1976. Anatomical, physiological and biochemical studies on the cerebellum from mutant mice. III. Protein differences associated with the weaver, staggerer and nervous mutation. Brain Res. 103:291–312 Mallouk R.S. 1975 Longevity in vertebrates is proportional to relative brain weight. Fed. Proc. 34: 2102–3 Mallouk, R.S. 1976 Author's reply (to: Aging in vertebrates: Allometric considerations of spleen size and lifespan by W.A. Calder III). Fed. Proc. 35: 97–98 Mangold–Wirz, K. 1966. Cerebralisation und Ontogenesemodus bei Eutherien. Acta Anat. 63:449–508 Mann, M.D., Towe, A.L. Glickman, S.E. 1986. Relationship between brain size and body size among Myomorph rodents. Soc. Neurosci Abs. 12:111 Mariani, J., Crepel,F., Mikoshiba, K., Changeux, J.P., and Sotelo, C. 1977. Anatomical, physiological and biochemical studies of the cerebellum from reeler mutant mouse. Phil. Trans. R. Soc. (Lond.) 281:1–28 Martin, R.D. 1981. Relative brain size and basal metabolic rate in terrestrial vertebrates. Nature 293: 57–60 Martin, R.D., Harvey, P.H. 1985. Brain size allometry. Ontogeny and Phylogeny. In Size and Scaling in Primate Biology. ed. W.L. Jungers. pp. 147–73. New York: Plenum Martini, E. 1912. Studien uber die Konstanz histologischer Elemente. III. Hydatima senta. Z. wiss. Zool. 102:425–645 Maurin, Y, Berger, B., Le Saux, F., Gay, M., Baumann, N. 1985. Increased number of locus ceruleus noradrenergic neurons in the convulsive mutant Quaking mouse. Neurosci. Lett. 57:313–18 Mayr, E. 1963. Animal Species and Evolution. Cambridge, Mass.: Belknap Press of Harvard Univ. Press McLoon, S., C. 1982 Alternation in precision of the crossed retinotectal projection during chick development. Science 215: 1418–20 Mellon Jr., D., Tufty, R.H., Lorton, E.D. 1976. Analysis of spatial constancy of oculomotor neurons in the crayfish. Brain Res. 109:587–94 Meyer, M.P., Morrison, P. 1960. Tissue respiration and hibernation in the thirteen–lined ground–squirrel, Spermophilus tridecemlineatus. Bull. Museum Comp. Zoo. 124:405–21 Miale, I.L., Sidman, R.L. 1961. An autoradiographic analysis of histogenesis in the mouse cerebellum. Exp. Neurol. 4:277–96 Mos, W., Williamson, R. 1986. A quantitative analysis of the spinal motor pool and its target muscle during growth in the Dogfish, Scyliorhinus canicula. J. Comp. Neurol. 248:431–40 Mullen, R.J. 1977 Site of gene action and Purkinje cell mosaicism in pcd<–>normal chimeric mice. Nature 270:245–47 Mullen, R.J., Eicher, E.M., Sidman, R.L. 1976 Purkinje cell degeneration. A new neurological mutant in the mouse. Proc. Natl. Acad. Sci. USA 73: 208–12 Mullen, R.J. and Herrup, K. (1979) Chimeric analysis of mouse cerebellar mutants. In: Neurogenetics: A Genetic Approach to the Central Nervous System. ed., X.O. Breakefield, pp. 271–97. New York: Elsevier–North Holland Muller, K.J., Nicholls, J.G., Stent, G.S. 1982. Neurobiology of the Leech. New York: Cold Spring Harbor Press Munk, O. 1966. Ocular degeneration in deep–sea fishes. Galathea Report, Scientific results of the Danish deep–sea expedition round the world 1950–52, 8:22–31 Narayanan, C.H., and Narayanan, Y. 1978. Neuronal adjustments in developing nucleus centers of the chick embryo following transplantation of an addition optic primordium. J. Embryol. Exp. Morph. 44:53–70 Newman, H.H., Patterson, J.T. 1911. The limits of hereditary control in armadillo quadruplets: A study of blastogenic variation. J. Morph. 22:855–926 Newport, J., Kirschner, M. 1982 a. A major developmental transition in early Xenopus embryos I. characterization and timing of cellular changes at the midblastula stage. Cell 30:675–86 Newport, J., Kirschner, M. 1982 a. A major developmental transition in early Xenopus embryos II. Control of the onset of transcription. Cell 30:687–96 Nicholson, J. L., Altman, J. 1972. The effects of early hypo– and hyperthyroidism on the development of rat cerebellar cortex. I. Cell proliferation and differentiation. Brain Res. 44:13–23 Nieto–Sampredro, J., Saneto, R.P., De Vellis, J., Cotman, C.W. 1985. The control of glial populations in brain: Change in astrocyte mitogenic and morphogenic factors in response to injury. Brain Res. 343: 320–28 Nordeen, E.J., Nordeen, K.W., Sengelaub, D.R., Arnold, A.P. 1985. Androgens prevent normally occurring cell death in a sexually dimorphic spinal nucleus. Science 229:671–673. Nordlander, R.H., Edwards, J.S. 1969. Postembryonic brain development in the monarch butterfly. II. The optic lobes. Wilhelm Roux Arch. Dev. Biol. 163:197–220 Ogawa, F. 1939. The nervous system of earthworm (Pheretima communissima) in different ages. Sci. Rep. Tohoku Univ. [Med]. 13:395–488. Okado, N., Oppenheim, R.W. 1984. Cell death of motoneurons in the chick embryo spinal cord. IX. The loss of motoneurons following removal of afferent input. J. Neurosci. 4: 1639–52 O'Leary, D.D.M., Fawcett, J.W., Cowan, W.M. 1986. Topographic targeting errors in the retinocollicular projection and their elimination by selective ganglion cell death. J. Neurosci. 6:3692–3705 Oppenheim, R.W. 1986. The absence of significant postnatal motoneuron death in the brachial and lumbar spinal cord of the rat. J. Comp. Neurol. 246: 281–286. Oppenheim, R.W. 1987. Cell death during neural development. In Handbook of Physiology, Neuronal Development, Vol 1. ed. W.M. Cowan. in press, Washington, D.C.: American Physiological Society (ultimately published in Ann. Rev. Neurosci. 1989 or 1990) Pakkenberg B, Gundersen HJG (1997) Neocortical neuron number in humans: effect of sex and age. J. Comp. Neurol. 384:312–320. Perkins, C.S., Aguayo, A.J., Bray, G.M. 1981. Schwann cell multiplication in Trembler mice. Neuropath. Appl. Neurobiol. 7:115–26 Pilar, G., Landmesser, L., Burstein, L. 1980. Competition for survival among developing ciliary ganglion cells. J. Neurophysiol. 43:233–254 Pond, C.M. 1977. The significance of lactation in the evolution of mammals. Evolution 31:177–99 Potten, C.S., Wichmann, H.E., Loeffler, M., Dobek, K., Major, D. 1982. Evidence for discrete cell kinetic subpopulations in mouse epidermis based on mathematical analysis. Cell Tiss. Kinet. 15: 305–29 Prestige, M.C. 1970. Differentiation, degeneration, and the role of the periphery: Quantitative considerations. In The Neurosciences, Second Study Program, F.O. Schmitt, ed. pp. 73–82. New York. Rockerfeller Univ. Press Provis, J., van Driel, D., Billson F.A., Russel, P. 1985. Human fetal optic nerve: Overproduction and elimination of retinal axons during development. J. Comp. Neurol. 238:92–101 Purves, D., Rubin, E., Snider, W.D., and Lichtman, J. 1986. Relation of animal size to convergence, divergence, and neuronal number in peripheral sympathetic pathways. J. Neurosci. 6:158–63 Quigley, H.A., Addicks, E.M., Green, W.G. 1982. Optic nerve damage in human glaucoma. III. Quantitative correlation of nerve fiber loss and visual field defect in glaucoma, ischemic neuropathy, papilledema, and toxic neuropathy. Arch. Ophthalmol. 100:135–46 Quinn, L.S., Holtzer, H., Nameroff, M. 1984 Age dependent changes in myogenic precursor cell compartment sizes. Evidence for the existence of a stem cell. Exp. Cell Res. 154: 65–82 Quinn, L.S., Holtzer, H., Nammeroff, M. 1985 Generation of chick skeletal muscule cells in groups of 16 from stem cells. Nature 313:692–94 Quinn, L.S., Nameroff, M. 1983a. Analysis of the myogenic lineage in chick embryos. III. Quantitative evidence for discrete compartments of precursors cells. Differentiation 24:111–23 Quinn, L.S., Nameroff, M. 1983b. Analysis of the myogenic lineage in chick embryos. IV. Effects of conditioned medium. Differentiation 24:124–30 Quinn, L.S., Nameroff, M. 1986. Evidence for a myogenic stem cell. In Molecular Biology of Muscle Development. UCLA Symposia on Molecular and Cellular Biology. New Series. Volume 29. eds. Emerson, C., Fischman, D.A., Nadal–Ginard, B., Siddiqui, M.A.Q., pp. 35–45. New York: Alan R. Liss Raff, M.C., Hornby–Smith, A., Brockes J.P. 1978. Cyclic AMP as a mitogenic signal for cultured rat Schwann cells. Nature 273:672–73 Rager, G. 1980 Development of the retinotectal projection in the chicken. Adv. Anat. Embryol. Cell Biol. 63: 1–92 Rakic, P. 1985. Limits of neurogenesis in primates. Science 227:1054–56 Rakic, P., Riley, K.P 1983a. Overproduction and elimination of retinal axons in the fetal rhesus monkey. Science 219:1441–44 Rakic, P., Riley, K.P. 1983b. Regulation of axon number in primate optic nerve by prenatal binocular competition. Nature 305: 135–137 Raymond, P.A. Reifler, M.J., Rivlin, P.K. and Clendening, B. 1986. Progenitor cells specific for rods lose their specificity in regenerating goldfish retina. Soc. Neurosci Abst. 12:118 Reh, T.A., Tully, T. 1986. Regulation of tyrosine hydroxylase–containing amacrine cell number in larval frog retina. Devel. Biol. 114:463–469 Ricklefs, R.E., Marks, H.L. 1984. Insensitivity of brain growth to selection of four–week body mass in Japanese quail. Evolution 38:1180–85 Rodieck, R.W., Brening, R.K. 1983. Retinal ganglion cells: Properties, types, genera, pathways and trans–species comparisons. Brain Behav. Evol. 23:121–164 Romanes, G.J. 1946. Motor localization and the effects of nerve injury on the ventral horn cells of the spinal cord. J. Anat. 80:117–31 Sacher, G.A., Staffeldt, E.F. 1974. Relation of gestation time to brain weight for placental mammals: Implications for the theory of vertebrate growth. Amer. Natur. 108:593–615. Salzer J.L., Bunge, R.P., Glaser, L. 1980. Studies of Schwann cell proliferation. III. Evidence for the surface localization of the neurite mitogen. J. Cell Biol. 88:767–78 Sanes, J.R., Rubenstein, J.L.R., Nicolas J–F. 1986. Use of a recombinant retrovirus to study post–implantation cell lineage in mouse embryos. EMBO J. 5:3133–42 Satorre, J., Cano, J., Reinoso–Surez, F. 1986. Quantitative cellular changes during postnatal development of the rat dorsal lateral geniculate nucleus. Anat. Embryol. 174:321–27 Schoenwolf, G.C. 1985. Shape and bending of the avian neuroepithelium: Morphometric analyses. Dev. Biol. 109:127–39 Shankland, M., Weisblat, D.A. 1984. Stepwise commitment of blast cell fates during the positional specification of the O and P cell lines in the leech embryo. Devel. Biol 106:326–42 Shariff, G.A. 1953. Cell counts in the primate cerebral cortex. J. Comp. Neurol. 98:381–400 Sholl, D. 1948. The quantitative investigation of the vertebrate brain and the applicability of allometric formulae to its study. Proc. R. Soc. Lond. 135:243–58 Shook, B.L., Maffei, L., Chalupa, L.M. 1984. Functional organization of the cat's visual cortex after prenatal interruption of binocular interactions. Proc. Natl. Acad. Sci. USA 82:3901–5 Sidman, R.L., Lane, P.W., and Dickie, M.M. 1962. Staggerer, a new mutation in the mouse affecting the cerebellum. Science 137:610–12 Silver, J., Hughes, A.F.W. 1974. The relationship between morphogenetic cell death and the development of congenital anophthalmia. J. Comp. Neurol. 157:281–302 Silver, J. 1978. Cell death during development of the nervous system. In Handbook of Sensory Physiology Vol. IX. ed. M. Jacobson. pp. 419–436. New York: Springer Verlag. Sonmez, E., Herrup, K. 1984. Role of staggerer gene in determining cell number in cerebellar cortex. II. Granule cell death and persistence of the external granule cell layer in young mouse chimeras. Dev. Brain Res. 12:271–283. Spitzka, E.A. 1903 Brain weights of animals with special reference to the weight of the brain in macaque monkey. J. Comp. Neurol. 13: 9–17 Stephan, H. 1958. Vergleichend–anatomische Untersuchungen an Insektivorengehirnen. Morphol. Jahrb. 99:853–80 Sternberg, P.W., Horvitz, H.R. 1984. The genetic control of cell lineage during nematode development. Ann. Rev. Genet. 18:489–524 Stewart, R.R., Spergel, D., Macagno, E.R. 1986. Segmental differentiation in the leech nervous system: The genesis of cell number in the segmental ganglia of Haemopis marmorata. J. Comp. Neurol. 253:253–59 Stewart, R.R., Gao, W.–Q., Peinado, A., Zipser, B., Macagno, E.R. 1987 Cell death during gangliogenesis in the leech: Bipolar cells appear and then degenerate in all ganglia. J. Neurosci. 7:1919–27 Stone, J. 1983. Parallel Processing in the Visual System. The Classification of Retinal Ganglion Cells and its Impact on the Neurobiology of Vision. Plenum Press, New York Storrs, E.E., Williams, R.J. 1968. A study of monozygous quadruplet armadillos in relation to mammalian inheritance. Proc. Natl. Acad. Sci. USA 60:910–14 [Strom, R.C., Williams, R.W. 1998. Cell production and cell death in the generation of variation in neuron number. J. Neurosci. In press. Aug98RW] Studnicka, F.K. 1905. Die Parietalorgane. In Lehrbuch der vergleichende mikroskopischen Anatomie der Wirbeltiere. A. Oppel, ed. vol. 5. S.G. Fischer Verlag, Jena. Sulston, J.E. 1976. Post–embryonic development in the ventral cord of Caenorhabditis elegans. Phil. Trans. R. Soc. Lond. Ser. B. 275:287–97 Sulston, J.E., Horvitz, H.R. 1977. Post–embryonic cell lineages of the nematode Caenorhabditis elegans. Devel. Biol. 56:110–156 Tanaka, H., Landmesser, L.T. 1986a. Interspecies selective motoneuron projection patterns in chick–quail chimeras. J. Neurosci. 6:2880–88 Tanaka, H., Landmesser, L.T. 1986b. Cell death of lumbosacral motoneurons in chick, quail, and chick–quail chimera embryos. A test of the quantitative matching hypothesis of neuronal cell death. J. Neurosci. 6:2889–99 Tåning, ÅA.V. 1950 Influence of the environment on number of vertebrae in teleostean fishes. Nature 165: 28 Taylor, B.A. 1971. Genetic relationships between inbred strains of mice J. Heredity 63:83–86 Temple, S., Raff, M.C. 1986 Clonal analysis of oligodendrocyte development in culture: Evidence for a developmental clock that counts cell divisions. Cell 44: 773–79 Thoenen, H. and Barde, Y.A. 1980. Physiology of nerve growth factor. Phys. Rev. 60:1284–1335 Thoenen, H., Korsching, S., Heumann, R., and Acheson, A. 1985. Nerve Growth Factor. Ciba Fndn. Symp. 116:113–28 Tower, D.B. 1954. Structural and functional organization of mammalian cerebral cortex: The correlation of neurone density with brain size. J. Comp. Neurol. 101:19–52 Torrence, S.A. 1983. Ascidian larval nervous system: Anatomy, ultrastructure, and metamorphosis. Ph.D. thesis, University of Washington, Seattle Trent, C., Tsung, N. Horvitz, H.R. 1983. Egg–laying defective mutants of the nematode Caenorhabditis elegans. Genetics 104:619–47 Truman, J.W. 1983. Programmed cell death in the nervous system of an adult insect. J. Comp. Neurol. 216:445–52. Truman, J.W. 1984. Cell death in invertebrate nervous systems. Ann. Rev. Neurosci. 7:171–88 Turner, D.L, Cepko, C.L 1987. A common progenitor for neurons and glia persists in rat retina late in development. Nature 328: 131–36 van Essen, D.C. Newsome, W.T., Maunsell, J.H.R. 1984. The visual field representation in striate cortex of the macaque monkey: Asymmetries, anisotropies, and individual variability. Vision Res. 24:426–48 van Essen, D.C., Newsome, W.T, Maunsell, J.H.R., Bixby, J.L. 1986. The projections from striate cortex (V1) to areas V2 and V3 in the macaque monkey: Asymmetries, areal boundaries and patchy connections. J. Comp. Neurol. 244:451–80. Vernon, J.A., Butsch, J. Effect of tetraploidy on learning and retention in the salamander. Science 125:1033–34. von Szily, A. 1912 Uber die einleitenden Vorgänge bei der ersten Entstehung der Nervenfasern im N. opticus. Albrecht von Graefe's Archiv. f. Ophthal. 81:67–86, plates 5 and 6. Waddington, C.J. 1942 Canalization of development and the inheritance of acquired characters. Nature 150: 563–65 Ware, R.W., Clark, D., Crossland, K., Russell, R.L. 1975 The nerve ring of the nematode Caenorhabditis elegans. Sensory input and more output. J. Comp. Neurol. 162: 71–110 Weisblat, D.A., Harper, G., Stent, G.S., Sawyer, R.T. 1980. Embryonic cell lineages in the nervous system of the Glossiphoniid leech Helobdela triserialis. Dev. Biol. 76:58–78. Weisblat, D.A., Sawyer, R.T., Stent, G.S. 1978. Cell lineage analysis by intracellular injection of a tracer enzyme. Science 202:1295–98. Weisblat D.A., Blair, S.S. 1982 Cell lineage in leech neurogenesis during normal development and after the ablation of identified blastomeres. NRP Bull. 20:783–93. Wetts, R., Herrup, K. 1982. Interaction of granule, Purkinje and inferior olivary neurons in lurcher chimeric mice. II. Granule cell death. Brain Res. 250:358–362. Wetts, R. and Herrup, K. 1983. Direct correlation between Purkinje and granule cell number in the cerebella of lurcher chimeras and wild–type mice. Dev. Brain Res. 10:41–47. White, J., Southgate, E., Thomson, J.N., Brenner, S. 1976 The structure of the ventral nerve cord of Caenorhabditis elegans. Phiolos. Trans. R. Soc. Lond. Ser. B 275: 327–48 Whitington, P.M., Bate, M., Seifert, E., Ridge, K., Goodman, C.S. 1982. Survival and differentiation of identified embryonic neurons in the absence of their target muscles. Science 215:973–75. Williams, R.W., Bastiani, M.J., Lia, B., Chalupa, L.M 1986. Growth cones, dying axons, and developmental fluctuations in the fiber population of the cat's optic nerve. J. Comp. Neurol. 246:32–69. Williams, R.W., P. Rakic 1988a. Elimination of neurons from the lateral geniculate nucleus of rhesus monkeys during development. J. Comp. Neurol. 272: 424–436. Williams, R.W., P. Rakic. 1988b. Three–dimensional counting: An accurate and direct method to estimate the number of cells in sectioned material. J. Comp. Neurol. 278: 344–52. Williams, R.W., Ryder, K., Rakic, P. 1987. Emergence of cytoarchitectonic differences between areas 17 and 18 in the developing rhesus monkey. Soc. Neurosci. Abstr. 13: 1044. [Williams, R.W. 2000. Mapping genes that modulate brain development: a quantitative genetic approach. In: Mouse brain development (Goffinet AF, Rakic P, eds). Springer Verlag, New York, pp 21Ð49.] Wilson M.A. 1971. Optic nerve fibre counts and retinal ganglion cell counts during development of Xenopus laevis (Daudin). Quart. J. Exp. Physiol. 56:83–91. Wimer, R.E., Wimer, C.C., Vaughn, J.E., Barber, R.P., Balvanz, B.A., Chernow, C.R. 1976. The genetic organization of neuron number in Ammon's horns of house mice. Brain Res. 118:219–43. Wingert, F. 1969. Biometrische Analyse der Wachstumsfunktionen von Hirnteilen und Körpergewicht der Albinomaus. J. Hirnforsch. 11:133–97. Winklbauer, R., Hausen, P. 1983. Development of the lateral line system in Xenopus laevis. III. Cell multiplication and organ formation in the supraorbital system. J. Embryol. Exp. Morphol. 76: 283–296 Wright, S. 1978. Evolution and the Genetics of Populations. Vol. 4: Variability within and among Natural Populations. The University of Chicago Press, Chicago Yablokov, A.V. 1974.Variability of Mammals. New Delhi, Amerind Publishing Co Young, J.Z. 1971. The Anatomy of the Nervous System of Octopus vulgaris. Oxford; Clarendon Press. Zamenhof, S. 1941 Stimulation of the proliferation of neurons by the growth hormone. I. Experiments on tadpoles. Growth 5:123–39 Zamenhof, S. 1942 Stimulation of cortical cell proliferation by the growth hormone. III. Experiments on albino rats. Physiol. Zool 15:281–92 Zamenhof, S. Mosley, J., Schuller, E. 1966. Stimulation of the proliferation of cortical neurons by prenatal treatment with growth hormone. Science 152:1396–97 Zamenhof, S. Mosley, J., Schuller, E. 1971. Prenatal cerebral development: Effects of restricted diet, reversal by growth hormone. Science 174: 954–55 Footnote 1. For a particular vivid example of the limitation of this single variable approach, see the controversy surrounding the correlation between brain size and longevity (Mallouk, 1975, 1976, Clader 1976). RETURN TO TEXT. Footnote 2. The only instance we are aware of in which an increase in neuron number may be maladaptive is the case of the mutant mouse quaking. Maurin et al (1985) have reported that the number of noradrenergic neurons in the locus coeruleus of quaking is about 20% above normal. This increase is associated with convulsion. RETURN TO TEXT. Footnote 3. This may be particularly true at early stages of development. As Martin (1981) has argued persuasively, the amount of energy that can be transferred from parent to offspring may set an upper limit to brain size. The advent of fully functional lactation in mammals before the end of the Triassic period was undoubtedly a key innovation that enabled mammals to sustain relatively large populations of neurons and glial cells (Pond 1977, Lillegraven 1979), and this in turn may have contributed significantly to the rapid radiation of mammals during the early Cenozoic. Speciation rates in mammalian genera are estimated to be about five times higher than rates in lower vertebrate genera (Bush et al 1977). The rate of growth of mammalian young that possess particularly large brains is often slow, and gestation and maturation take a long time (Sacher & Staffeldt 1974). We see this pattern clearly in primates (Gould 1977, p. 367), but it is also apparent in other mammals. For instance, the nectar-feeding bat, Glossophaga sorichina, has a very high brain/body weight ratio, and Eisenberg (1981, p. 307) points out that the young may develop slowly principally because the milk contains little fat. This strategy allows the mother to defray the cost of building a new brain over a longer period. In general, animals with big brains have small litters, slow development, and intense parental care. RETURN TO TEXT. Footnote 4. Hamburger (1975) has pointed out, however, that the
overproduction of spinal motoneurons cannot be ascribed to an imprecise
programming of the number of mitotic cycles. He observed that the total
number of spinal motor neurons produced before the onset of neuron death
in a number of 5.5- and 6-day chicken embryos was relatively constant.
(range from 18,900 to 21,600, n = 11).
RETURN TO TEXT. Since 11 August 98
|
Neurogenetics at University of Tennessee Health Science Center
| Print Friendly | Top of Page |
Mouse Brain Library | Related Sites | Complextrait.org
