Note to the reader: This is a revised edition of a paper published in Developmental Medicine and Child Neurology (2000;42:652–662). The definitive original print version is copyright ©2000 by Developmental Medicine and Child Neurology.
New figures, text, and links have been incorporated into the revision. Revised html (http://nervenet.org/netpapers/Rosen/Strength2000/Strength.html) copyright ©2001 by Glenn D. Rosen
SINGLE CAUSE, POLYMORPHIC NEURONAL MIGRATION DISORDERS: AN ANIMAL MODEL
Glenn D. Rosen and Albert M. Galaburda
Dyslexia Research Laboratory and Charles A. Dana Research Institute, Beth Israel Deaconess Medical Center; Division of Behavioral Neurology, 330 Brookline Avenue, Boston MA 02215; Harvard Medical School, Boston, MA 02115.
Address Correspondence to:
Glenn D. Rosen
Department of Neurology
Beth Israel Deaconess Medical Center
330 Brookline Ave
Boston, MA 02215
USA
Email: grosen@caregoup.harvard.edu
Aim: Injury to the developing cortical plate can result in variety of neuronal migration disorders. We report the results of experimental research aimed at determining whether these different types of neocortical malformations are the consequence of comparable injury of varying intensity.
Method: We placed freezing probes on the skulls of newborn rats (equivalent to 4–5 month of gestation in humans) and induced either one or two freezing injuries of durations ranging from 2–20 s.
Results: A variety of cortical malformations including minor laminar dysplasias, molecular layer ectopias, microgyria, and porencephalic cysts were seen in the brains of these animals. The severity of the malformation was directly related to the strength (number of hits and duration) of the freezing injury.
Conclusion: These results suggest that a single etiologic event of varying severity during neuronal migration to the neocortex can induce widely disparate malformations of the cortex.
Neuronal migration disorders are structural malformations of the cerebral cortex, cerebellum or hippocampus and include heterotopias, microgyria, porencephaly, lissencephaly, and pachygyria (1–3). These malformations have been associated with a wide variety of developmental clinical disorders (4), including intractable epilepsy (5–8), dyslexia (9,10), hemiparetic cerebral palsy (11,12), congenital muscular dystrophy (13–16), and developmental delay (17,18). The etiology of these malformations have been reviewed elsewhere (19–21), and it is clear that they can result from genetic or epigenetic events occurring during the period of neuronal migration. In general, neuronal migration disorders of genetic etiology are more diffuse, whereas those associated with injury or other epigenetic events are more focal. For example, about 90% of individuals with Miller-Dieker syndrome have microdeletions in a critical 350-kilobase region in chromosome 17p13.3 (LIS1, 22,23). Additional genes have been associated with X-linked lissencephaly and double cortex syndrome (doublecortin, 24–27). In contrast, it is generally accepted that microgyria and other focal disorders of neuronal migration are the result of secondary effects of injury occurring during critical periods of development (28–33).
Animal models have been developed for many of these neuronal migration disorders. The reeler mouse, for example, has a cortex that is characterized by an inversion of the normal laminar pattern (34–39), and is genetically modulated by reelin gene products (40,41). A similar mouse mutation, scrambler (42), appears to be modulated by a genes (disabled-1, mdab-1) the products of which act downstream of reelin (43,44). A mutant strain of rats (tish) form an ectopic collection of neurons below the cortical plate, which appears to be neocortical in origin and morphology (45,46). These animals are susceptible to spontaneous seizures, and the tish animals are proposed to be a model for human double cortex.
Malformations can be induced in otherwise normal rodents through a variety of mechanisms. Rats injected prenatally with methylazoxymethanol (MAM) have microcephalic brains and ectopic neurons in the subcortical white matter and the CA1 region of the hippocampus (47–49). These rats have a decreased seizure threshold (50,51), and changes in glutamate receptors. Injection of neurotrophin-4 into the ventricles of developing rats causes the formation of diffuse heterotopias in the molecular layer (layer I) of the neocortex (52). Focal molecular layer ectopias can be induced in by puncture wounds at or around birth (53) or by exposure to gamma irradiation in utero (54–56). Finally, a malformation resembling human four-layered microgyria can be induced by hypoxic/ischemic damage to the developing cortical plate by either freezing injury (57–62) or by ibotenic acid injections (63,64) during the period of late neuronal migration. Slices of microgyric cortex have been shown to be epileptogenic (65–68), and male rats with microgyria have been shown to have difficulties similar to those seen in individuals with dyslexia in the ability to process fast auditory information (69,70).
The advent of these animal models has made possible the exploration of key factors involved in the ontogeny of the cerebral cortex, as well enabling a better understanding of the mechanisms involved in the development of the malformations themselves. One feature of human neuronal development that is not completely understood is the co-occurrence of multiple malformations in the same brain (e.g., 71). For example, polymicrogyria and/or pachygyria has been reported in association with white and gray matter heterotopias (72–75). In some severe cases porencephaly, polymicrogyria, and heterotopias were seen together, and it was suggested that they might all result from a single etiologic event (76,77). Yet, it could be that different kinds of injuries acquired at the same time of gestation could each cause a separate type of malformation (59). Finally, the same etiological event, albeit acting at the same time, may vary in severity and lead to different malformations.
In this experiment, we tested the hypothesis that different types of malformations could be induced by injury to the developing cortical plate delivered at the same age but with increasing severity. Specifically, we exposed individual neonatal rats to either one or two freezing injuries ranging in duration from 2 to 20s and categorized the resultant malformations of the cortex.
Pregnant Wistar rats were obtained from Charles River Laboratories (Wilmington, MA) on gestational day 16–18 (E16–18). On the day after birth (P1), animals were randomly assigned to receive either one or two freezing injuries to the left hemisphere or a sham surgery. Cortical development of P1 rats is equivalent to that of humans at 4–5 months of gestation. Those receiving freezing injury were further assigned an injury duration of either 2, 5, 10, or 20 s. On P2, P21 or P60, the animals were perfused transcardially with 0.9% saline followed by 4% paraformaldehyde, and their brains subsequently processed for immunohistochemistry and standard histology
Induction of Microgyria
Microgyria were induced based on a modification of a technique by Dvorák and colleagues (57,58), and reported in detail elsewhere (61,62). Pups were anesthetized with hypothermia, and a small incision was made in the anteroposterior plane of the skin over the midline, exposing the skull. For those receiving a single injury, a cooled (-70°C) 2 mm diameter stainless steel probe was placed on the skull over the left hemisphere, approximately 2 mm lateral of the sagittal suture and 2 mm caudal of bregma, for either 2, 5, 10, or 20 s. Those receiving two injuries were treated identically, except that the first was placed 1 mm caudal of bregma and the second 1 mm rostral of lambda. Animals receiving sham surgery were treated identically to those receiving freezing injury except that the probe was maintained at room temperature. After surgery, the skin was quickly sutured, subjects were marked with identifying ink injections to the footpads, warmed under a lamp, and returned to their mother.
Histology
For all perfusions, subjects were anesthetized (P2 by hypothermia; P21 or P60 by pentobarbital [60 mg/kg i.p.]) and were transcardially perfused with 0.9% saline followed by 4% paraformaldehyde. The brains were removed from the skulls and placed into fresh 4% paraformaldehyde for 24 h. The brains were then placed in a 10% sucrose solution in 0.1 M sodium phosphate buffer for at least 24 hr, and then placed in 30% sucrose buffer until the brains sank. A nick was made on the ventral surface of the right hemisphere, and the brains were frozen on dry ice, serially sectioned in the coronal plane at 40 µm, and the sections stored in 0.1 M sodium phosphate buffer. One series of every tenth section was stained with Thionin for Nissl substance (P2, P21, and P60). An adjacent series was stained for neurofilament (P21 and P60 only) as this stain had previously been shown that disruption of neurofilament-positive fibers followed neonatal freezing lesions (61), and further that this disruption could provide complementary information concerning the extent of the malformation.
Nissl
Mounted sections were stained for Nissl substance with 0.05% Thionin using standard techniques.
Neurofilament Immunohistochemistry
Free-floating sections were rinsed twice in phosphate buffer saline (PBS; pH 7.4) for five min each and transferred to a buffered 0.6% hydrogen peroxide solution in order to block staining of endogenous peroxidases. The sections were rinsed twice in PBS and incubated overnight at 4°C in a 1/1000 dilution of an antibody cocktail to the 68 kDa and 150 kDa subunits of neurofilament (SMI-311; Sternberger Monoclonals Inc., Baltimore, MD). The vehicle (diluent) was 3% rabbit serum in PBS with 0.3% Triton X-100
Sections were then placed into a solution containing the linking antibody (rabbit anti-mouse immunoglobulin; Dakopatts (Santa Barbara, CA) Z259 - diluted 1/20) at room temperature for two h. The sections were rinsed twice with PBS and placed in a 1/250 dilution of mouse peroxidase anti-peroxidase (Dakopatts B650) at room temperature for two h. The tissue was rinsed twice in PBS, twice in 50 mM Tris buffer (pH 7.6), and developed using 0.05% diaminobenzidine and 0.005% hydrogen peroxide diluted in Tris. After rinsing with Tris, sections were mounted on chrome-alum coated slides, dehydrated, counterstained with Methyl Green/Alcian Blue, and coverslipped with Permount
Analysis
All sections were analyzed under light microscopy and the extent and distribution of cortical malformation noted and documented with photomicroscopy.
Acute Damage
Previous research had demonstrated that 24 hours following a single 5 s freezing injury at P1 a distinct, focal region of necrosis underlying the freezing probe appeared (62). In the current experiment, variable amounts of cortical necrosis were apparent at P2 that were directly proportional to the length of exposure to the freezing probe. Thus, a single 2 s freezing injury resulted in disturbance to the superficial portion of the cortical plate but led to little or no damage to the deeper portions (Fig. 1A). As the duration of the freezing injury increased, the extent of the necrotic area expanded in all dimensions centrifugally from the area of probe placement—rostro-caudal, medio-lateral, and superficial-deep (Fig. 1). For injury durations of greater than 2 s, the entire cortical plate was destroyed. In the case of the 20 s freezing injury, evidence of necrosis down through the subplate and the white matter was noted.
 |
Figure 1 - A. Photomicrograph from a P2 rat subjected to a 2 s freezing injury on P1. Note relatively sparse area of necrosis in the cortical plate (arrowheads). B. Photomicrograph from a P2 rat subjected to a 5 s freezing injury on P1. Note area of necrosis (arrowheads), which is much larger than seen following a 2 s freezing injury (panel A), and encompasses the entire cortical plate. C. Photomicrograph from a P2 rat subjected to a 10 s freezing injury on P1. Note area of necrosis (arrowheads), which is larger in both medio-lateral plane and in the dorso-ventral plane than seen following freezing injuries of shorter duration (panels A,B). Arrows denote some sparing of the subplate cells, although not complete. D. Photomicrograph from a P2 rat subjected to a 20 s freezing injury on P1. Note area of necrosis (arrowheads), which is far larger in both medio-lateral plane and in the dorso-ventral plane than seen following freezing injuries of shorter duration (panels A,B,C). Necrosis includes the entire cortical plate and the subplate cells. White arrowhead points to necrosis extending into the white matter. Bar = 200 µm for each panel.
Abbreviations: cp = cortical plate, sp = subplate cells, wm = white matter |
Single Injury Groups
Brains were examined at P21 or at P60. There were no differences in the appearance of lesions at those two ages, therefore their data were pooled. Cortical malformations were noted in all experimental groups and in none of the shams. For clarity, each of the eight injury groups are reported separately below.
2 seconds (N=4)
Animals receiving a single 2 s injury had the least severe form of cortical malformation. Typically, these were characterized by regions of microdysgenesis extending a relatively small distance into the cortex (Fig. 2A). These microdysgenetic areas showed occasional buckling of the molecular layer, but there was rarely evidence of a fused molecular layer such as that seen in typical microgyria (Fig. 2B). In those cases where microgyria were seen, the depth of the microsulcus extended only to layer IV. Molecular layer ectopias were occasionally noted at the rostral and caudal tips of the malformation, and there were also cell free regions in throughout the dysgenetic cortex. Neurofilament-stained sections showed some drop-off in immunoreactivity where the small buckling appeared (Fig. 2C).
5 seconds (N=5)
In our previous work, all microgyria were induced by a single 5 s freezing injury, and the results of the animals in the present experiment closely resembled the previously reported findings. Specifically, the most common type of malformation was a microgyrus characterized by the infolding of superficial cortical layers to form a microsulcus with opposed, fused pial surfaces, which varied in depth from 50–80% of the thickness of the neocortex. The predominant orientation of the microsulcus was in the rostral-caudal plane. The molecular layer lined the microsulcus in its entirety, although its thickness varied. Layers II and III were invariably preserved wholly or in part, and swept deep toward the region of neuronal loss, paralleling the course of the microsulcus (Fig. 2D,E). Effectively, therefore, this freezing injury recreated the common microgyric four-layer cortex comprising (i) an infolded molecular layer, (ii) a superficial (infolded) neuronal layer (corresponding to ordinary layers II, III but less layered), (iii) a neuron-free zone containing the astroglial remnants of the injured cortical plate, (iv) a deep neuronal layer (corresponding to ordinary layer VIb). Molecular layer ectopias were occasionally present in the periphery of the lesion.
Neurofilament-immunoreactivity was absent from the regions of microdysgenesis as reported previously (61). Relatively minor and vague dysgenetic changes seen in Nissl stains were often quite easily delineated on neurofilament-stained sections (Fig. 2F).
10 seconds (N=7)
Microgyria were the most common cortical malformation seen following a single 10 s freezing injury. In contrast to 5 s injuries, the microsulci had a greater depth (75–90% of the cortical mantle), sometimes distorting the underlying white matter. Occasionally, two microsulci would flank a small gyrus (microgyrus). The rostro-caudal extent of the malformation was also greater following these longer durations (Fig. 2G,H). At the periphery of the lesion, molecular layer ectopias were also seen.
As with the 5 s injury, neurofilament-immunoreactivity was absent in the regions of malformation (Fig. 2I).
20 seconds (N=4)
As expected, a single 20 s freezing injury resulted in a more severe malformation than that seen following injury of shorter durations. Specifically, microsulci often extended entirely to the white matter, thereby resulting in a malformation without a visible layer iii sweeping underneath layer ii of the malformation. As with the 10 s injury, a second microsulcus often formed, thereby causing a microgyrus (Fig. 2J,K). As with the previously described animals, molecular layer ectopias were seen at the periphery of the malformation.
As with the 5 s and 10 s injury groups, neurofilament-immunoreactivity was absent in the regions of microdysgenesis (Fig. 2L).
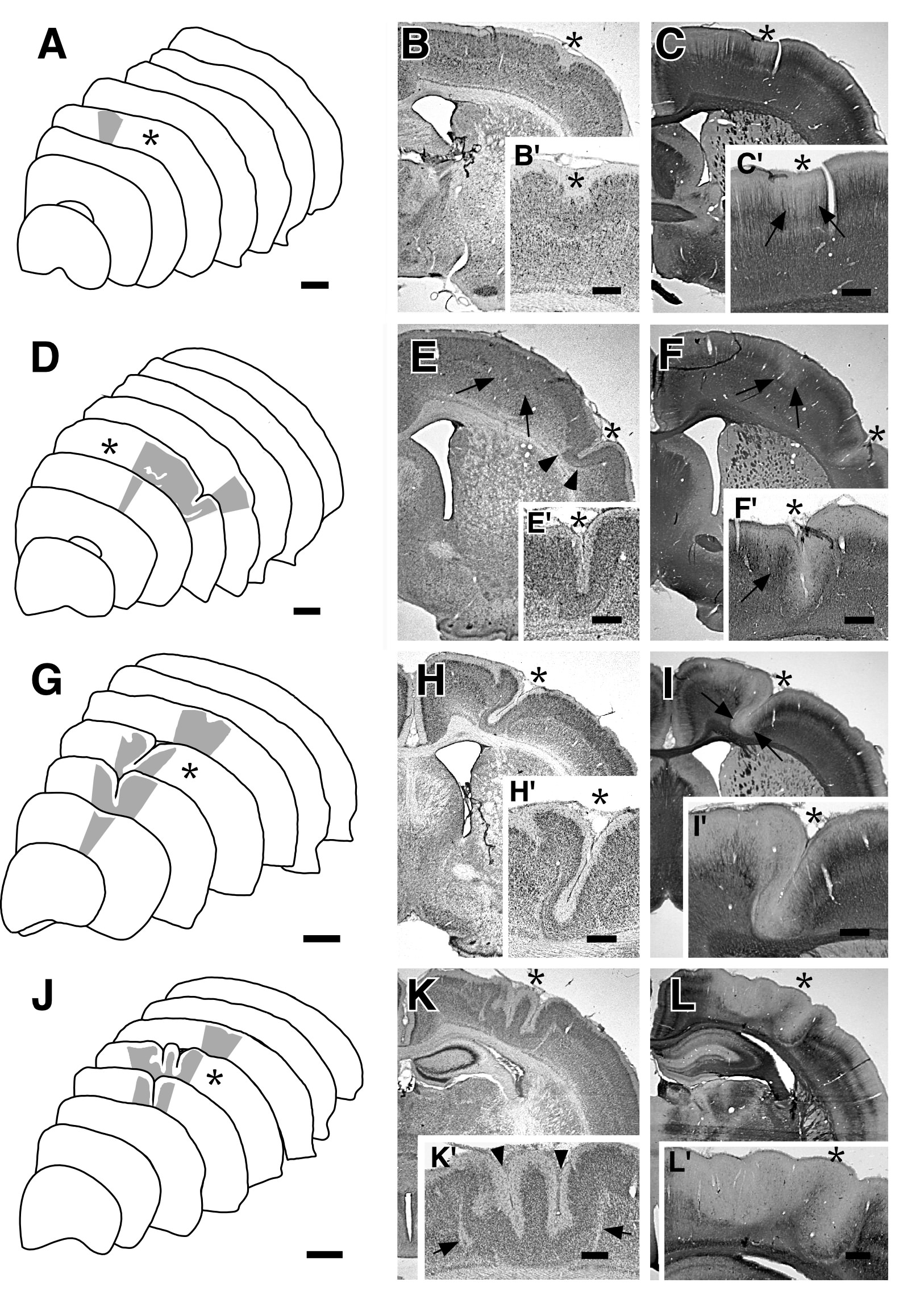 |
| Figure 2 - A. Tracings from left hemisphere of a rat sacrificed at P21 following a single 2 s freezing lesion to the cortical plate at P0. Grayed region denotes area of microdysgenesis. Asterisk denotes section photomicrographed in accompanying panels. Bar = 1 mm. B. Nissl-stained section showing region of microdysgenesis (asterisk). B'. Higher power magnification showing microdysgenetic region. Note deflection of the molecular layer (asterisk). Bar for B = 800 µm, bar for B' = 400 µm. C. Section slightly rostral to B immunohistochemically stained for neurofilament. Note the slight decrease in neurofilament staining in the region of the damage in both C (asterisk) and C' (asterisk and arrows). Bar for C = 800 µm, bar for C' = 400 µm. D. Tracings from left hemisphere of a rat sacrificed at P21 following a single 5 s freezing lesion to the cortical plate at P0. Grayed region denotes area of microdysgenesis. Asterisk denotes section photomicrographed in accompanying panels. Bar = 1 mm. E. Nissl-stained section showing region of microgyria (asterisk) with cell free region (arrowheads). There is region of less severe microdysgenesis medially which is not as evident with Nissl stains (arrows) E'. Higher power magnification of microgyria. Bar for E = 800 µm, bar for E' = 400 µm. F. Section adjacent to E immunohistochemically stained for neurofilament. Note the decrease in neurofilament staining in the region of the microgyria (asterisk) and in the medial dysgenetic area (arrows). F'. Higher power photomicrograph of microgyric region (asterisk). Note preservation of neurofilament-immunoreactive fibers on the medial portion of the microgyrus (arrow). Bar for F = 800 µm, bar for F' = 400 µm. G. Tracings from left hemisphere of a rat sacrificed at P21 following a single 10 s freezing lesion to the cortical plate at P0. Grayed region denotes area of microdysgenesis. Asterisk denotes section photomicrographed in accompanying panels. Bar = 1 mm. H. Nissl-stained section showing region of microgyria (asterisk). H'. Higher power magnification of microgyria. Bar for H = 800 µm, bar for H' = 400 µm. I. Section adjacent to H immunohistochemically stained for neurofilament. Note the decrease in neurofilament staining in the region of the microgyria (arrows). I'. Higher power photomicrograph of microgyric region (asterisk). Bar for I = 800 µm, bar for I' = 400 µm. J. Tracings from left hemisphere of a rat sacrificed at P21 following a single 20 s freezing lesion to the cortical plate at P0. Grayed region denotes area of microdysgenesis. Asterisk denotes section photomicrographed in accompanying panels. Bar = 1 mm. K. Nissl-stained section showing malformation (asterisk). K'. Higher power magnification of malformation. Note the two microsulci (arrowheads) forming a microgyrus. Arrows denote cell-free regions (layer iii). Bar for K = 800 µm, bar for K' = 400 µm. L. Section caudal to B immunohistochemically stained for neurofilament. Note the decrease in neurofilament staining in the region of the malformation (asterisk). L'. Higher power photomicrograph of malformation (asterisk). Bar for L = 800 µm, bar for L' = 400 µm. |
Double Injury Groups
In each experimental group, two freezing injuries to the developing cortical plate led to more severe malformations than their single injury counterparts. As with the single injury groups, neurofilament-immunoreactive fibers were mostly absent in the malformations. The results for each injury group are summarized below.
2 seconds (N=3)
Two distinct regions of malformation were noted following two 2 s freezing injuries. Each of these areas of malformation was similar in appearance to those seen following single injury of a similar duration, although somewhat more severe. Specifically, these malformations were characterized mostly by laminar dysplasias and molecular layer ectopias but regions of microgyria were also noted. When present, these microgyric regions were composed of microsulci that extended to 20–50% of the depth of the cortical mantle (Fig. 3A).
5 seconds (N=5)
All of the brains exposed to two 5 s freezing injuries had microgyria. There was a wide variability in the appearance of the malformations, however. In 2 cases, two distinct microgyria were seen in the rostral and caudal portions of the brain, each one similar in appearance to a microgyrus following a single injury. In each of the remaining three brains, there were only single malformations that were most likely the result of a fusing of the rostral and caudal lesions, as the rostro-caudal extent of the anomaly was much greater than that seen after a single 5 s injury. Further, the severity of the malformation following two freezing injuries was greater than that seen following a single injury, as evidenced by the depth of the microsulcus, which reached the white matter (Fig. 3B).
10 seconds (N=2)
In both cases, the malformations following two 10 s injuries were very severe, consisting mostly of microgyria with very deep microsulci. In one case, the two lesions fused into one malformation that ran almost the entire rostro-caudal extent of the hemisphere. In this case, the malformation impinged on the white matter below, clearly deflecting, and perhaps impeding, the passage of fibers. There was, in addition, local ventricular enlargement noted in the caudal portions of the malformation. In the second case, distinct rostral and caudal malformations were seen. Both of these malformations had deep microsulci and showed severe distortions of cortical lamination (Fig. 3C)
20 seconds (N=5)
Two freezing injuries of 20 s duration resulted in very severe malformations. These malformations consisted of molecular layer ectopias at the periphery of the malformation, microgyria, and (in three cases) porencephalic cysts. In all cases there was fusion of the rostral and caudal lesions and the malformation generally extended for most of the rostro-caudal extent of the neocortex (Fig. 3D). Local ventricular dilatation was seen in all subjects, and hippocampal degeneration and thalamic degeneration (especially the MGN) was noted in 4 of the subjects (Fig. 4).
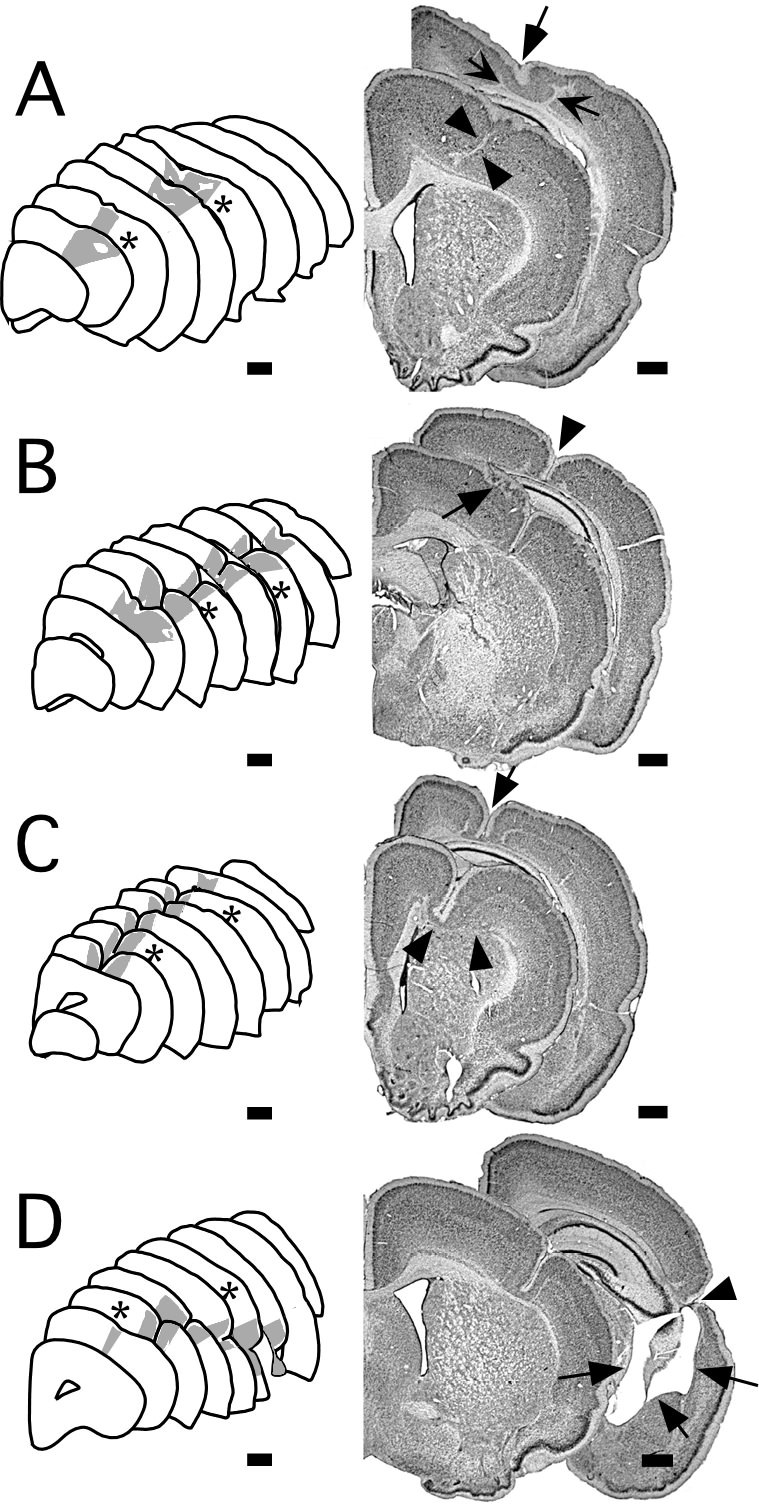

These results demonstrate that a single type of injury delivered to the cortical plate during late neuronal migration can induce a wide variety of cortical malformations—ranging from molecular layer ectopias, through cortical dysplasias, through microgyria, to porencephalic cysts—depending upon the severity of the injury. Thus, molecular layer ectopias and minor cortical dysplasias were associated with a single 2 s freezing injury. As the duration of freezing injury increased, additional types of malformations (mostly microgyria) were seen. The most severe injury, two 20-s exposures, resulted in porencephalic cysts. It is important to note that the more severe malformations were often seen in conjunction with the more minor malformations. For example, molecular layer ectopias were seen following injuries strong enough to induce microgyria, as we and others have demonstrated previously (57,61,66). Further, microgyria and molecular layer ectopias were seen following the induction of porencephalic cysts with two 20 s injuries (Fig. 5).
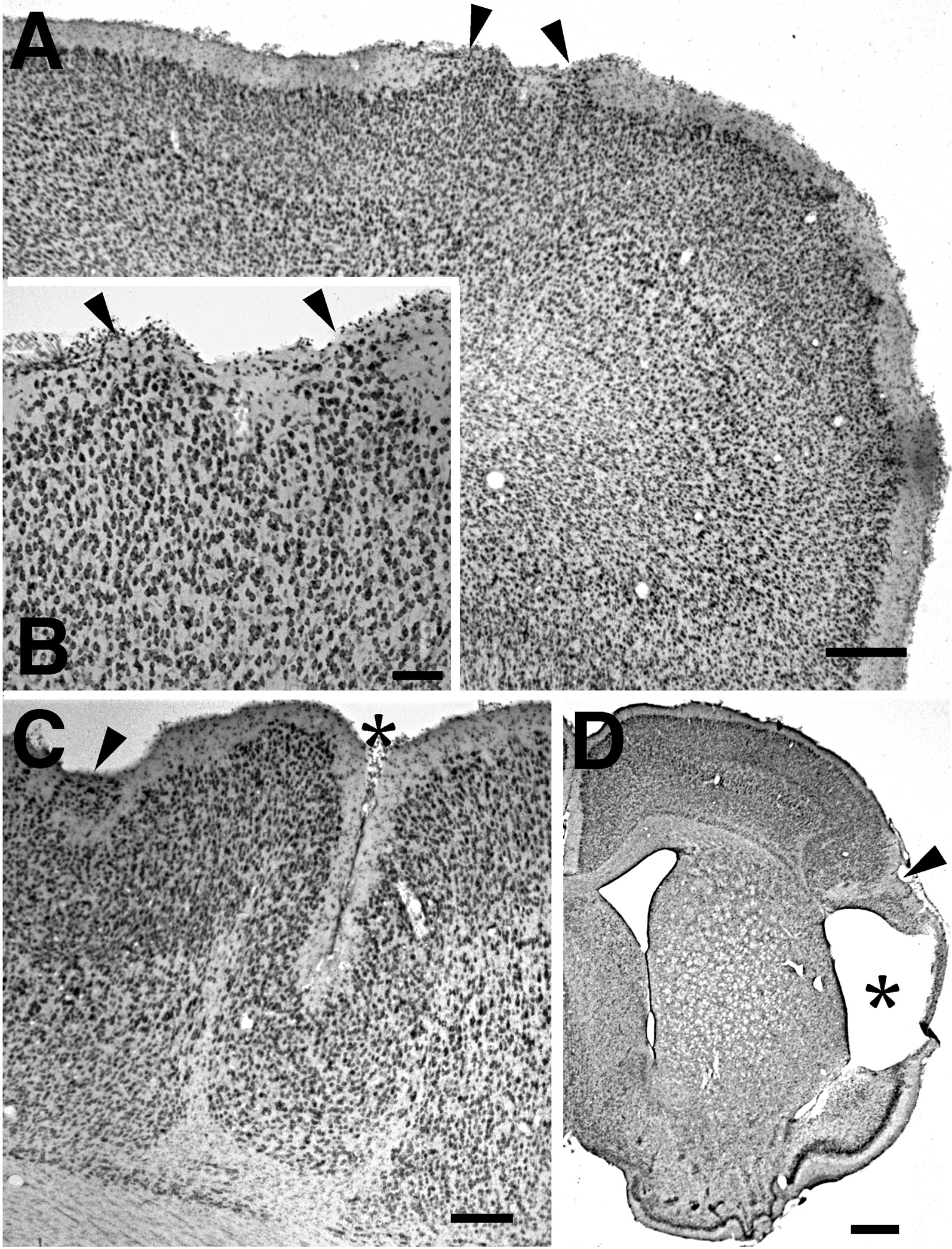 |
Figure 5 - A. Photomicrograph showing ectopic collections of neurons in the molecular layer of a P21 rat subjected to two 20 s freezing lesions on P1 (arrowheads). Bar = 400 µm. B. Higher power photomicrograph of molecular layer ectopias seen in panel A (arrowheads). Bar = 100 µm. C. Photomicrograph from a more caudal section from the same animal in Panels A and B showing molecular layer ectopia (arrowhead) in conjunction with a microgyrus (asterisk). Bar = 200 µm. D. Photomicrograph showing porencephalic cyst (asterisk) in association with microgyria (arrowhead) in a P21 rat subjected to a two 20 s freezing lesions on P1. This is a different animal than seen in Panels A-C. Bar = 800 µm. |
These findings raise a number of questions. What is the mechanism of action of these freezing injuries so that they cause these types of malformations? What changes to the developing brain occur following injuries of varying severity that would result in malformations of such disparate appearance? How can different types of malformations appear in the same brain?
Mechanism of action of freezing injuries
We have previously reported a breach in the external glial limiting membrane (EGLM) associated with formation of neocortical ectopias in immune-disordered mice (78). This breach in the EGLM can be seen as early as E15 in the mouse and we have seen that neurons originally destined to each of the cortical layers migrate through this breach to take their position in the molecular layer. We have also demonstrated that a puncture wound delivered to the cortical plate acts to mechanically disrupt the EGLM and also leads to the formation of molecular layer ectopias (53). The importance of the EGLM to the formation of these types of anomalies has been recognized by others as well (31,60). Marin-Padilla, in his study of 33 human cases, found that subpial hemorrhagic injury resulted in the breakdown of the EGLM. Further, in the course of the brain’s response to the injury, molecular layer ectopias were formed. Periventricular hemorrhages occurring at the same gestational age, on the other hand, did not result in molecular layer ectopias, but rather caused the cessation of neuronal migration altogether (31). Thus, similar injuries affecting different regions of the developing brain can cause profoundly different malformations.
The mechanism for the formation of microgyria following early freezing injury is not yet known, although clues can be derived from the study of changes in the brain that follow the injury (60,62). Neuronal necrosis can be observed as soon as 1.5 h after a single 5 s freezing injury. At the same time, radial glial fibers in the area begin to disappear and there are a few reactive astrocytes in the area of the lesion. Interestingly, the pial surface is intact. One day after the injury, there is total necrosis of the cortical plate, sparing only layer VIb (or subplate) neurons. Radial glial fibers have completely withdrawn from the area of damage, but their cell bodies appear to be intact. Excess numbers of macrophages appear in the damaged area, and there is a dramatic increase in the number of reactive astrocytes. Three days post-injury, radial glial fibers regrow though the region of intense astrogliosis, and neurons normally destined for layer II–III of the cortex (79) begin to migrate through that area. There is a marked increase in the number of macrophages present, and the size of the area of tissue necrosis decreases. The emergent form of microgyria can clearly be seen by day 5, starting primarily at the periphery of the lesion. Radial glial fibers continue their regrowth through the damaged area, and some GFAP-positive fibers adopt the appearance of radial glia. One week after the injury, there are large numbers of radial glial-like fibers stained by GFAP in the malformation. The microgyria gain their adult appearance by P15, at which time radial glial fibers can still be seen in the area of damage, although not elsewhere. These histologic changes indicate that microgyria are the outcome of basic brain repair mechanisms following a presumably hypoxic/ischemic injury occurring during the end of the period of neuronal migration (60,62,80,81).
We have seen that with increased exposure of the cortical plate to the freezing probe the extent of cortical necrosis increases (Fig. 1). Thus, a 2 s freezing injury shows a cone of necrosis that does not progress entirely through the cortical plate but destroys the EGLM and some superficial neurons. The depth of the necrosis increases proportionally with duration of the insult such that two 20 s freezing injuries result not only in the complete destruction of the cortical plate, the subplate cells, but perhaps also the subjacent proliferative ventricular zone as well. We hypothesize that the destruction of this proliferative zone results in the cessation of neuronal and glial migration to the portion of the cortical plate (31, discussed above). Thus deprived of neurons and glia, the injured cortex undergoes cystic necrosis and forms a porencephalic cyst that connects to the ventricular space. Alternatively, it could be that the zone of necrosis following two 20 s freezing injuries is so extensive that it blocks migration of neurons and glia to the cortex. If it were a question of mechanical blockage of migration, however, one would expect these stymied neurons to accumulate elsewhere (presumably subcortically) and the resultant malformation should more closely resemble periventricular neuronal heterotopia. We have seen no evidence of this type of malformation in our cases.
Multiple Malformations in the Same Brain
There were a number of cases where microgyria were present along with neocortical ectopias, and porencephalic cysts were always seen in association with microgyria. The hypothesized relationship between severity of cortical necrosis and the resultant malformations discussed above may also explain the coexistence in some cases of two or more types (Fig. 6). In cases where two or more types of malformation occurred in the same brain, the less severe one always appeared at the periphery of the lesion. We suggest that this is because the greatest depth of tissue necrosis takes place in that portion of the cortex directly underlying the freezing probe. Areas surrounding the center were injured less deeply, as identified by the cone-shaped area of necrosis following a freezing injury (62).
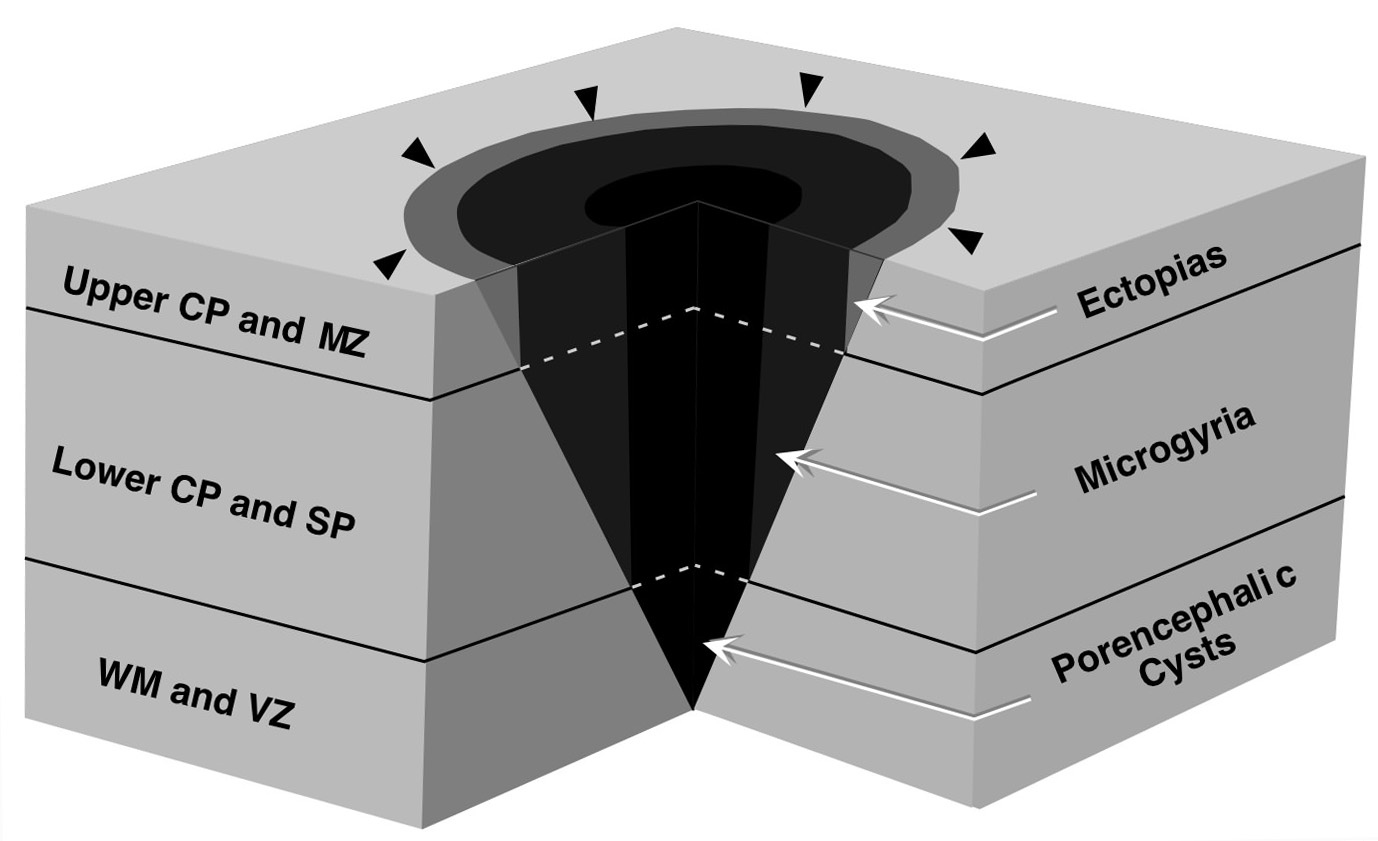 |
Figure 6 - Schematic illustrating one scenario for the co-occurrence of different types of malformations from the same lesion. In this example, a freezing probe is placed on the cortical surface of a developing rat. In the center of the probe, which is the coldest part (dark gray), the cortical plate (CP), subplate (SP), white matter (WM), and ventricular zone (VZ) are damaged. The damage becomes progressively less severe as one moves radially outward from the center of the probe so that at the periphery (light gray), only the marginal zone (MZ) and upper CP are damaged. In regions where only the upper CP and MZ are damaged, then ectopias are likely to form. When the damage extends into the lower CP and SP (gray), then microgyria-like malformations will be noted. When the damage extends into the WM and VZ, then porencephalic cysts will result. |
Conclusions
The results of the current study support the notion that a variety of cortical malformations can be caused by a single etiologic event occurring at the same time during late neuronal migration in the rat. What looks like dramatically different malformations is simply the result of differences in the severity of the injury. Thus, in the situation in which the damage to the developing cortex is less severe, either because the duration (short) or the location (periphery) of the freezing lesion, molecular layer ectopias are seen. When the injury is more severe because of proximity and/or duration, microgyria and ectopias follow. With the most severe injury, porencephaly, microgyria, and ectopias are seen.
The mechanism for the infolding, distortion and rupture of the cortex is not clear, but is appears to involve at least in part graded damage to the cortical plate, migrating neurons and glia, and germinal zones. Research on the chemical, mechanical, and molecular events surrounding early damage to the cortical plate will yield further insights into the creation of these malformations. It is hoped that insight gained from these and other animal models of induced malformations (63,64,82) will allow a better understanding of the pathogenesis of the wide range of human developmental disorders that are associated with disorders of late neuronal migration, such as those mentioned in the introduction. Specifically, the understanding of the biology underlying the formation of these types of developmental malformations may be useful for the prevention, diagnosis and management of certain learning disabilities and epilepsies of childhood.
ACKNOWLEDGEMENTS
This work was supported, in part, by grant HD 20806 from the Public Health Service of the USA. The authors wish to acknowledge the technical support of Alison Frank.