Note to the reader: This is a revised edition of a
paper published in Journal of Neuropathology and Experimental Neurology (1991;50:145–160).
The definitive original print version is copyright ©2000 by The American Association of Neuropathologists.
New figures, text, and links have been incorporated into the revision. Revised HTML (http://nervenet.org/netpapers/Rosen/MG91/MG.html) copyright ©2001 by Glenn
D. Rosen
FREEZING LESIONS OF THE DEVELOPING RAT BRAIN: A MODEL FOR CEREBROCORTICAL MICROGYRIA
Peter Humphreys,1 Glenn.D. Rosen, Douglas M. Press, Gordon F. Sherman, and Albert M. Galaburda
Dyslexia Research Laboratory and Charles A. Dana Research Institute, Department of Neurology, Beth Israel Deaconess Medical Center; Harvard Medical School, Boston, MA.
1Section of Neurology, Children’s Hospital of Eastern Ontario, 401 Smyth Road, Ottawa, Ontario K1H 8L1.
Correspondence to:
Glenn D. Rosen, Ph.D.
Department of Neurology,
Beth Israel Deaconess Medical Center
330 Brookline Ave.
Boston, MA 02215
Email: grosen@caregroup.harvard.edu
Cerebrocortical microgyria were induced by placing a freezing probe on the skull of P0 and P1 rat pups. Freezing lesions resulted in laminar necrosis of the infragranular layers and the subsequent migration of supragranular neurons through the region of damage. The end result was most often a region of four-layered microgyric cortex consisting of a molecular layer, a thickened layer ii, a lamina dissecans (corresponding to the necrotized layers IV, V, and VIa), and a neuronal layer iv which corresponded to layer VIb of the intact cortex. Immunocytochemical investigation of the microgyric cortex with antibodies to neurofilament, glial fibrillary acidic protein and glutamate showed more widespread disruption of neocortical architecture than could be seen from Nissl appearance. In contrast, vasoactive intestinal peptide-containing neuronal bodies appeared to be distributed normally in the microgyric region although their processes were sometimes distorted. These results are considered in the light of previous research on induced microgyria, and possible implications for the behavioral consequences of focal developmental neuropathology are discussed.
Micropolygyria has been observed in various human neuropathologic disorders including porencephaly (1–3), thanatophoric dysplasia (4), microencephaly (5), parabiotic twin syndrome (6), and occasionally in developmental dyslexia (7,8). The abnormality can be very focal or can involve a major portion of the cerebral convexity.
The pathogenesis of four-layered microgyria, the most common type, has been the subject of speculation since its first description. Bielschowsky (9,10) discussed several mechanical notions of earlier workers—related to size inbalance between grey and white matter leading to increased folding—and proposed his own explanation that micropolygyria was the result of a fundamental disorder in neuronal migration, a hypothesis that has since been discarded.
The role of injury is now widely accepted (3,11–13), and more recent debates have focused on the timing of the origin of the malformation, i.e., whether microgyria originates during or after completion of neuronal migration to the neocortex. Some workers have advocated the former since “microdysgenesis” (minor abnormalities in the formation of 6-layered cortex which include nests of neurons and glia in the molecular layer neuronal ectopias, ectopic neurons in subjacent white matter, and focal irregularities in arrangement of cortical neuronal layers) were seen at the edges of the malformation (12,14,15). Critics of this hypothesis have shown in fetuses that the malformation may occur after completion of the period that is ordinarily considered to be the end of neuronal migration, i.e., after 20 to 24 weeks of gestation, and have suggested that the subcortical ectopic nests could be artifacts of section plane and the microdysgenetic foci could also be produced later in ontogenesis (3,13,16). Additional support for the postmigrational hypothesis has been adduced from Golgi studies where neurons in the upper layers of microgyria were not distinguishable from those in the upper laminæ normal cortex (17).
On the other hand, experimental work in rodents has tended to support the notion that the malformation occurs during neuronal migration, at least in this species (18,19). In those studies focal microgyria were produced in the rat brain by applying a freezing probe to the skulls of newborn rat pups. In the rat, neuronal migration continues for two or three days after birth, making the developmental chronology in the newborn animal roughly equivalent to the 18–24 gestational week-old human fetus. A sequential analysis of the evolving cortical freezing lesion suggested that the four-layered cortical lesion resulted from: (a) Cold-induced selective neuronal necrosis of the neocortical layers IV, V, and VIa, sparing cortical layer VIb, thus producing a relatively neuron-free zone corresponding to microgyric cortical layer III, and a neuron-dense zone (former layer VIb) corresponding to microgyric cortex layer IV; and (b) the subsequent migration of putative supragranular neurons through the zone of destruction to form that which was originally intended as cortical layers II, III and was now microgyric cortex layer II (Fig. 1). (Microgyric cortex layer I corresponded to the normal molecular layer (I) of 6-layer cortex). When a cluster of such freezing lesions was produced, a more extensive cortical lesion resulted with an appearance similar to that of so-called status verrucosus deformis of the human brain, having the appearance of neuronal migration abnormalities.
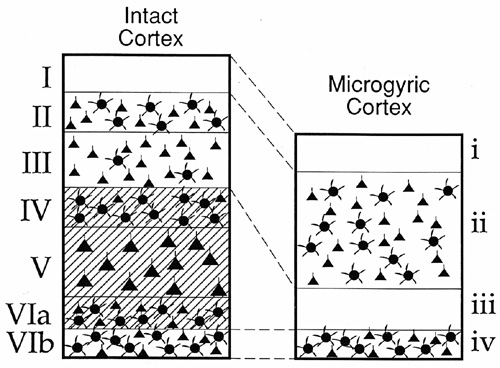 |
Figure 1 - Schematic illustrating the result of freezing lesions on P0 or P1 in the newborn rat pup on the eventual laminar arrangement of neurons in the adult rat neocortex. In the case of the intact cortex, there are six layers of cells denoted by uppercase letters. During the process of freezing, layers IV,V, and VIa are damaged. The resultant microgyric cortex (lower case letters) has four layers where layer iii is an area devoid of neurons and layer ii is larger than layer II and III of the intact cortex. |
Until now, no attempt has been made to repeat these experiments nor to determine whether there are more widespread disturbances of neocortical architecture associated with this apparently focal developmental neuropathology as seen in the more minor malformation of cerebrocortical microdysgenesis (20–22). In this paper we report results that replicate and extend the previous findings. These results support the observation that microgyria in the rat can be produced by injury during neuronal migration to the cerebral cortex, and further, that a more widespread disturbance in local neocortical architecture than noted in cell stains is present, probably the result of postmigrational maturational changes. The question of whether microgyria can also result from injury after the completion of neuronal migration to the neocortex, at least in the rat, will be addressed in a subsequent publication.
MATERIALS AND METHODS
Protocol
Time-mated Wistar rats (Charles River Laboratories, Wilmington, MA) were obtained on day 16 of gestation. On the day of birth (P0) or P1, rat pups were anesthetized and a freezing probe was placed directly on the skulls overlying the left hemisphere. Additional animals were subjected to a freezing lesion on P10 (after completion of neuronal migration to the neocortex), and the results will not be presented here. Littermate controls were not disturbed. The animals were then split into two groups—one for descriptive neuroanatomy (n = 41) and one to receive surgical intervention for connectional studies (to be discussed in a subsequent paper).
Animals were killed on postnatal days 21 and 60 under deep anesthesia (Pentobarbital) by intracardiac perfusion of 50 ml 0.9% saline followed by either 200 ml 10% formalin (for standard histological stains; n = 8) or by 500 ml 2% paraformaldehyde/0.05% glutaraldehyde (for immunocytochemistry; n = 33). Following perfusion, the formalin-perfused brains were removed from the skulls and placed in 10% formalin for at least 48 hours (h), then transferred to a 30% sucrose/10% formalin solution for a week or until the brain sank to the bottom of the jar. In most cases, the tissue was embedded in albumin-gelatin with a nick placed over the right hemisphere, frozen on dry ice, serially sectioned in the coronal plane at 30µm, and stored in 2% formalin. In a few cases, the brain was not embedded prior to sectioning. The paraformaldehyde/glutaraldehyde-perfused brains were removed from the skulls and placed in a 10% sucrose 0.1M sodium phosphate buffer for at least 24 h and then placed in 30% sucrose 0.1M sodium phosphate buffer until the brains sank. A nick was made on the ventral surface of the right hemisphere, and the brains were frozen on dry ice, serially sectioned in the coronal plane at 30 µm, and stored in 0.1M sodium phosphate buffer. One series of every tenth section was stained with Thionin for Nissl substance. Adjacent sections from the formalin-perfused brains were stained for myelin using the Loyez method, and for glial fibers using the phosphotungstic acid hematoxylin (PTAH) method. In the brains processed for immunocytochemical techniques, adjacent series were stained for neurofilament (NF; 68 kD subunit), vasoactive intestinal peptide (VIP), glial fibrillary acidic protein (GFAP), and glutamatergic neuronal processes (Glut) for the purpose of addressing specific disturbances of immunoarchitecture of the neocortex involved. Specifically, neurofilament was chosen in order to view in greater detail the neurons and dendrites in the regions surrounding the freezing lesion in the hope of seeing more widespread disturbances of neocortical architecture (22). Vasoactive intestinal peptide was chosen for analysis as previous research has demonstrated local changes in the distribution of VIP in and around regions of spontaneous neuronal ectopias (21). Furthermore, VIP has been shown to be relatively resistant to anoxic/ischemic insults (23). Glutamate, an excitatory neurotransmitter, is the primary neurotransmitter for cortical pyramidal neurons. Because glutamatergic neurons are especially vulnerable to hypoxic/ischemic insults (24,25), Glut is an ideal candidate for examination in this study. Glial fibrillary acidic protein was chosen to further explore for the presence of glia in the freezing lesion.
Sections were examined under low and high power light microscopy.
Induction of Focal Necrotic Lesions
Focal necrotic lesions were induced in the cerebral cortex of 1 day-old Wistar rat pups obtained from timed pregnancies using a modification of the technique employed by Dvorák and colleagues (18,19). Using hypothermia-induced general anesthesia, a small incision was made in the antero-posterior plane, in the skin over the left cerebral hemisphere, with exposure of the skull. A freezing probe (see below) was applied to the convexity of the skull approximately midway between bregma and lambda, for precisely timed periods ranging from 2–10 seconds. The skin was then quickly sutured, the animal warmed under a lamp, and then returned to the mother. Littermate controls were not given a freezing lesion.
The freezing probe consisted of a stainless-steel rod with one end tapered to give a precise contact point of 2 mm diameter. The rod was fixed inside a plastic centrifuge tube with the tip of the rod extruding through the bottom of the tube, fastened in place with a rubber grommet. The other end of the rod passed through the screw cap of the centrifuge tube, also fastened with a grommet. A second grommet in the cap admitted a thermometer probe to the interior of the centrifuge tube. The centrifuge tube was filled with (liquid) methylbutane and cooled by immersion in either liquid nitrogen or crushed dry ice. With liquid nitrogen the methylbutane was allowed to reach a temperature of -100°C before the freezing whereas with dry ice the maximum cooling which could be achieved was -70 to -75°C. The exposed tip of the stainless-steel rod was used to create the freezing lesions (Fig. 2).
The optimum exposure for P0,P1 freezing lesions was found to be 5 seconds when dry ice was used for cooling, while 2 seconds was sufficient to induce significant parenchymal necrosis using liquid N2. The P0,P1 freezing lesions reported here were induced with 5 seconds CO2 exposure.
 |
Figure 2 - Diagram of freezing apparatus used for the induction of freezing lesion on the skull of newborn rat pups. See text for further description. Bar = 10 mm |
Histological Preparation
Formalin-Fixed Sections
Nissl. Mounted sections were stained for Nissl substance with 0.05% Thionin using standard techniques.
Loyez. The free-floating sections were washed in distilled water for 30 seconds and placed for six h in a 2% ferric ammonium sulfate solution. The sections were then washed for 30 seconds and incubated in a 1% hematoxylin solution in 10% ethanol/2% lithium carbonate solution overnight at room temperature. The next day the sections were washed twice for 30 seconds each and differentiated in 2% ferric ammonium sulfate until the gray matter appeared. The sections were then washed in three changes of distilled water for 30 seconds each before differentiation in Weigert’s solution (2% sodium borate/2.5% potassium ferricyanide) solution. The sections were then washed three times in distilled water, with the second wash containing a few drops of ammonium hydroxide, before being mounted onto subbed slides, dehydrated, cleared with xylene, and coverslipped with Permount.
PTAH. The mounted sections were washed twice with distilled water for 30 seconds each and placed into a 1% iodine/2% potassium iodide solution for five minutes (min). After two distilled water washes for 30 seconds each, the sections were placed for five min in 95% ethanol. The sections were then placed in distilled water for 30 seconds, placed in a 0.25% potassium permanganate solution for five min, washed again for 30 seconds, and bleached in 5% oxalic acid for five min. After five washes in distilled water of 30 seconds each, the sections were stained with a 5% solution of phosphotungstic acid in hematoxylin for 24 h. The sections were then dehydrated, cleared, and coverslipped with Permount.
Immunocytochemical Stains
For all antibodies, a control series was run with the omission of the primary antibody. In addition, preimmune sera from the same species generating the primary antibody were used as controls. In each case negative staining was seen.
Neurofilament. Free-floating sections were rinsed twice in phosphate-buffered saline (PBS; pH 7.4) for five min each and transferred to a buffered 0.6% hydrogen peroxide solution in order to block staining of endogenous peroxidases. The sections were rinsed twice in PBS and incubated overnight at 4°C in a 1/50 dilution of mouse anti-neurofilament immunoglobulin (monoclonal antibody to the 68 kDa subunit of neurofilament from Boehringer Mannheim, Indianapolis MN). The vehicle (diluent) for all antibody incubations was 3% rabbit serum in PBS.
Sections were then placed into a solution containing the linking antibody (rabbit anti-mouse immunoglobulin - Dakopatts (Santa Barbara, CA) Z259 - diluted 1/20) at room temperature for two h. The sections were rinsed twice with PBS and exposed to a 1/250 dilution of mouse peroxidase anti-peroxidase (Dakopatts B650) at room temperature for two h. The tissue was rinsed twice in PBS and then twice in 50 mM Tris buffer (pH 7.6) and developed using 0.05% diaminobenzidine and 0.005% hydrogen peroxide diluted in Tris. After rinsing with Tris, sections were mounted on chrome-alum coated slides, dehydrated, counterstained with Methyl Green/Alcian Blue, coverslipped with Permount.
Vasoactive Intestinal Peptide (VIP). Initial preparation was identical to that for NF. Prior to the first antibody incubation, sections were placed in vehicle only for 20 min at room temperature. Sections were placed into a 1/2000 solution of primary antibody (Incstar, Stillwater, MN) overnight at 4°C. The vehicle (diluent) for all antibody incubations was 5% goat serum in PBS. The next day, sections were transferred into a biotinylated goat anti-rabbit immunoglobulin solution (Vector Laboratories, Burlingame, CA) diluted 1/60 for two h at room temperature. After two washes in PBS, the sections were placed into ABC complex (Vector Laboratories) for two h at room temperature. The tissue was rinsed twice in PBS and twice in 50 mM Tris buffer (pH 7.6) and developed, dehydrated, counterstained, and coverslipped as with NF.
Glutamatergic processes. The procedure of immunocytochemical staining of glutamatergic fibers is identical to that for VIP with the exception of the dilution of the primary antibody (Incstar) which is 1/5000 and a different biotinylated antibody in a different vehicle, horse anti-mouse IgG (Vector Laboratories).
Glial Fibrillary Acidic Protein. The procedure of immunocytochemical staining of GFAP is identical to that for VIP with the exception of the dilution of the primary antibody (Incstar), which is 1/25.
RESULTS
Lesion Production and Location
Consistent necrosis was induced uniformly by a 5 second exposure with a probe temperature of -70°C to 75°C. While freezing lesions were produced in every case, the location of the lesion within the left hemisphere was less predictable. Even though the freezing probe was placed over the central regions of the hemispheric mid-convexity (corresponding to underlying sensorimotor cortex) in each case, the resulting cortical lesion varied in location from frontal polar cortex to lateral auditory or even visual cortex. It is possible that the diameter of the probe tip (2mm) was too large for the small skull size in the neonatal rat, and that a 1 mm diameter probe would have produced a more consistently located, if smaller, neocortical lesion. Alternatively, individual differences in vascular supply may have contributed to the topographic variability, as we have considered that a vascular mechanism for the injury is important in this case (see below).
Description of Freezing Lesion at P0, P1
Since there were no significant differences in the appearance of lesions examined at P21 or P60, the lesions of animals in these groups will be described together.
The mildest, most subtle type of neocortical lesion consisted of a zone of neuronal loss and gliosis within layer V; layers II–IV, and VI appeared entirely normal. More severe lesions consisted of a much larger area of selective neuronal loss and the presence of glial nuclei involving layers IV, V, and VIa while Layer VIb was usually preserved. With this type of lesion there was characteristically an infolding of the superficial cortical layers to form a microsulcus with opposed, fused pial surfaces. The microsulcus varied in depth from 10–75% of the thickness of the neocortex. The molecular layer (I) followed the microsulcus in its entirety, although layer thickness varied. Layers II and III were invariably preserved in their whole or in part, and swept downward toward the region of neuronal loss, following the course of the microsulcus (see Fig. 3). The anterior-posterior extent of this type of lesion was 1.8–2.1 mm. Effectively, therefore, this most common type of freezing lesion recreated a microgyric four-layer cortex comprising (i) a molecular layer, (ii) a superficial (infolded) neuronal layer (corresponding to ordinary layers II, III but less layered), (iii) a neuron-free zone (necrotic area), (iv) a deep neuronal layer (corresponding to ordinary layer VIb).
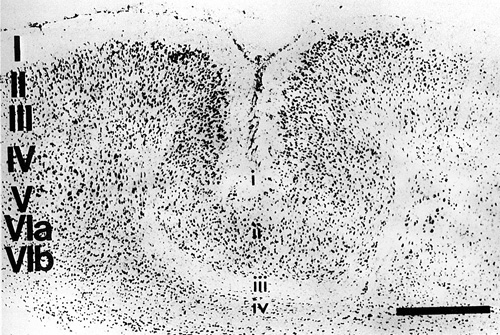 |
Figure 3 - Thionin-stained low power photomicrograph of region of typical neocortical microgyria in an adult rat following freezing lesion to the skull at P0. The layers of adjacent cortex medial to the lesion are shown at the left in upper case letters, and the resultant 4-layered microgyria is labeled in lower case letters. See Figure 1 and text for explanation. Bar = 500 µm |
In some cases, part of the freezing lesion led to two diverging microsulci which, in appropriate sections produced a true microgyrus sandwiched between the microsulci. Depending on the plane of section, the microgyrus sometimes appeared as a warty excrescence on the cortical surface. However, unlike the true status verrucosus deformis, cells did not reach the pial surface in these protuberances (Fig. 4).
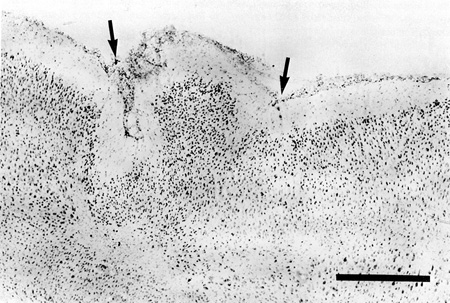 |
Figure 4 - Nissl-stained section showing two diverging microsulci (arrows) providing a warty excrescence at the pial surface which resembles a microgyrus in an adult rat subjected to a freezing lesion at P0. Bar = 500 µm |
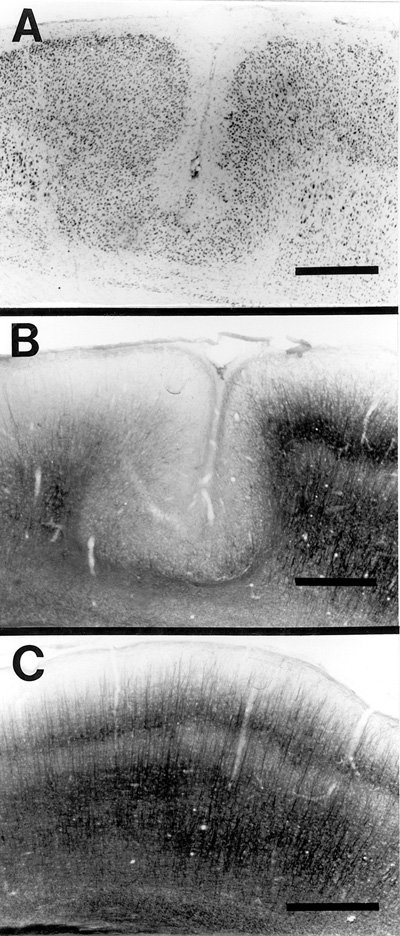
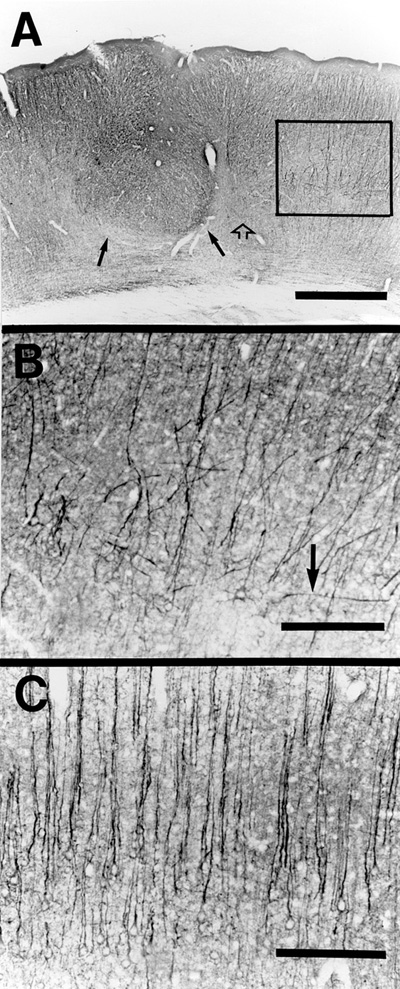
There was a generalized disruption of neurofilament staining, which oftentimes resulted in a complete absence of neurofilament stain within the area of the lesion. Specifically, there was a marked reduction in neurofilamentous staining in the portions of layers ii in the depth of the microsulci (Fig. 5a,b) when compared to layers II/III of normal controls (Fig. 5c). This may well be the result of the necrosis of layer V, which is normally the site of dense neurofilamentous staining such that the apical dendrites of layer V cells may provide the bulk of layer II, III staining. Sections stained for glutamatergic processes showed a marked diminution of glutamatergic processes in the zone of neuronal loss and in adjacent later VIb. Layer V zones immediately adjacent to the area of neuronal loss showed distorted placement/orientation of glutamatergic processes. This is in contrast to the orderly radial distribution of normal glutamatergic fibers (Fig. 6a, b, c).
 |
 |
|
| Figure 5 - A. Thionin stained region of typical neocortical microgyria in an adult rat following freezing lesion to the skull of at P0. B. Section adjacent to (A) immunocytochemically stained for neurofilament. Note disruption of the normal pattern of neurofilament distribution layer ii of the microgyria as compared to layer II/III of intact cortex (medial and lateral to the lesion). C. Neurofilament-like immunoreactivity in a the same region as (A) and (B) of a control (unlesioned) brain. Bar = 500 µm for all panels | Figure 6 - A. Glutamatergic-like immunoreactivity of the brain of an adult rat with microgyria induced by freezing lesions at P0. Solid arrows mark microgyria layer iii (lamina dissecans), and open arrow indicates regional absence of glutamatergic-like reactivity. Boxed area shows region enlarged in (B). Bar = 500 µm. B) Boxed region in (A) showing distortion of the normal pattern of glutamatergic-like immunoreactive fibers in the area of the freezing lesion. Arrow denotes fiber traversing perpendicular to the pial surface. Bar = 250 µm. C) Normal pattern of glutamatergic-like immunoreactivity in non-lesioned brain. Note density and radial alignment of fibers in comparison with (B). Bar = 250 µm |
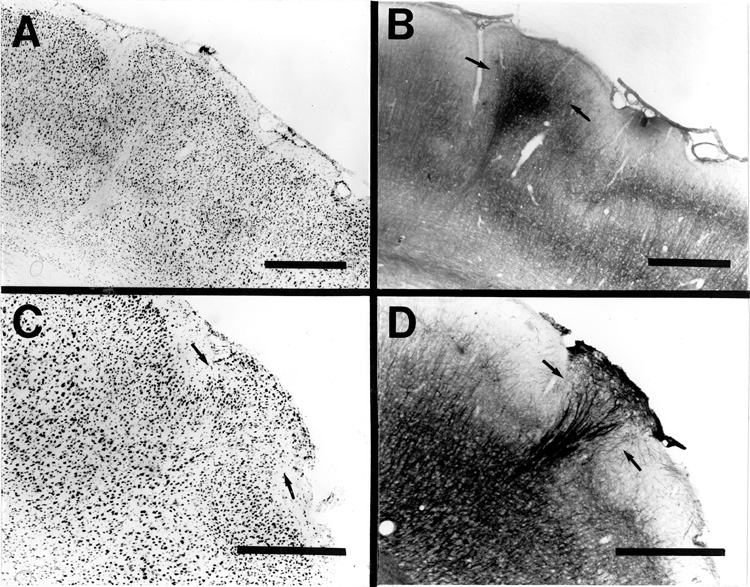
In the periphery of Nissl-stained freezing lesions, at sites in which the pial contour drastically changes direction toward becoming infolded, clusters of ectopic neurons were frequently observed in the molecular layer adjacent to the microsulcus (Fig. 7a). These often reached the pial surface and resembled the type of abnormality seen to occur spontaneously in immune-disordered mice (26,27) and in several individuals with developmental dyslexia (7, 8), and which we have termed cerebrocortical microdysgenesis (15; Fig. 7c). Additionally, identical nests of ectopic neurons in layer I have been noted in human microgyria (12). Increased presence of pial vessels were often also seen at these sites. Neurofilament staining of adjacent sections showed patterns of fiber staining in the induced ectopia (Fig. 7b) remarkably similar to those seen in the immune-disordered mice (Fig. 7d).
 |
|
Figure 7 - A. Nissl-stained section showing clusters of ectopic neurons in layer I of the induced microgyric cortex of an adult rat. These types of anomalies generally appear at the periphery of the lesion and look similar to those seen in the New Zealand Black (NZB) mouse (C). B. Neurofilament-stained section adjacent to (A) demonstrating similar patterns of fiber staining to that seen in the NZB mouse (D). Bar = 500 µm (all panels).
|
Freezing lesions of sufficient severity to disrupt layer VIb typically led to myelin staining in the zone of selective neuronal loss and adjacent sections stained for glial fibers by the PTAH method showed small foci of enhanced glial staining within the zone of neuronal loss. In addition, sections stained for GFAP showed clusters of GFAP-positive fibers within the zones of neuronal loss (Fig. 8a,b,c). These findings, taken together, indicate myelinated scarring in the area of the freezing lesion, also a characteristic of ulegyria in human developmental neuropathology in which early acquired lesions, before intracortical myelogenesis is complete, are felt to attract myelination (28,29).
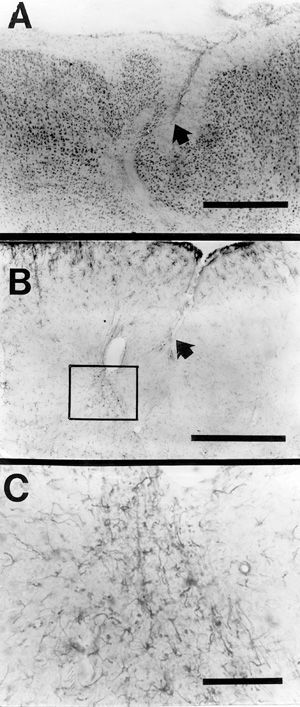 |
| Figure 8 - A. Nissl-stained section showing an area of microgyria in an adult rat. Arrow is for orientation with (B). Bar = 500 µm. B. Section reacted for GFAP. Note clusters of glial cells in the area of the microsulcus (box). Bar = 500 µm. C. Boxed area of (B) showing GFAP-positive cells. Bar = 90 µm. |
The distribution of VIP neuronal somata appeared relatively normal, with the distribution maintaining its laminar distribution in spite of the distortion caused by the freezing lesion and only occasional distorted and truncated processes in the area of damage.
Pathogenesis of Induced Microdysgenesis
The results of our studies confirm the findings of Dvorák and colleagues (18,19). It is clear that a freezing probe applied to the intact skull of the newborn rat pup, while the cerebral cortex is passing through the later states of neuronal migration, reliably produces a focus of four-layered cortex that substantially resembles the microgyric cortex seen in humans with a variety of neurologic dysfunctions (12,13). However, since neuronal migration to the upper layers proceeds relatively undisturbed after the injury, it is possible that injury after these neurons have completed their migration would also produce a microgyrus as it does prior to completion of migration. In other words, the issue of whether neurons are migrating or not may be irrelevant to the production of a microgyrus, assuming that the degree of injury to the deeper layers is severe enough during a time in which maturation is still ongoing. This question is currently under investigation in the rat model in this laboratory.
The pathogenesis of the microgyric lesion appears to be initially vascular (18). Intense cold transmitted through the calvarium from the freezing probe is thought to induce coagulation and thrombosis of adjacent pial vasculature. Occlusion of developing perforating cortical arterioles in turn results in tissue necrosis in the underlying cortex, which at this stage of development consists only of layers IV, V, and VI. That layer VIb neurons are often preserved probably reflects the fact that this layer is less dependent on perfusion from pial vessels (30). Migrating young neurons within the day 0 rat cortex—putative supragranular neurons—may be spared by the ischemic event because they have smaller metabolic requirements than mature neurons (31). In addition, these young migrating neurons may derive some protection from their attachment to the radial glia, which may be relatively preserved as evidenced by the persistent migration of supragranular neurons. The [3H]-thymidine data of Dvorák and Feit (18) suggest that it is the migrating young neurons from both the injured cortex and the intermediate zone that then travel through the necrotic zone and form a neuronal layer superficial to it, contiguous with layers II, III of the intact adjacent cortex.
The upper layers appear fairly normal in this malformation, except for the relative lack of differentiation between what would ordinarily be layers II and III. The morphology of the neocortical freezing lesion clearly reflects the continuation, albeit somewhat abnormal, of migration of supragranular neurons following the production of focal cortical necrosis. However, it is unlikely that a primary disorder of neuronal migration has actually occurred in this model. Thus, many layer II neurons wind up in layer II and layer III neurons at a slightly deeper level. Golgi studies indicate that the orientation of the axonal and dendritic processes of the supragranular neurons in the reconstituted neuronal layer external to the necrotic zone are, for the most part, appropriate (19).
Our results indicate that a disturbance of neuronal migration—consisting of nests of supragranular neurons in the molecular layer reaching up to the pia—has taken place at the periphery of freezing lesions (the lips of the microsulcus). Even here, however, rather than a primary error in migration, we postulate that damage to the molecular layer by excessive stretching during the formation of the microsulcus and/or damage by the plethora of penetrating pial vessels during the repair period has permitted some upper layer neurons to migrate beyond their natural barrier.
Regional Disturbances in Neuronal Architecture
Our immunocytochemical studies of the freezing lesions suggest that regional changes in post-migrational neuronal organization may be a far more important consequence of a P0,P1 rat neocortical lesion than a minor alteration in neuronal migration. Neurofilament-containing cell processes (axons and dendrites) were markedly reduced in the external cell layer of the microsulcus, even though the neuronal cell bodies appeared normal on Nissl stains. Without EM studies we cannot say whether the lack of NF staining reflects a deficiency in processes intrinsic to supragranular neurons themselves, or a deficiency of afferent axonal and dendritic processes from elsewhere (e.g., apical dendrites from layer V pyramids). The lack of NF staining is so striking that both intrinsic axodendritic and afferent axonal processes are probably involved.
On the periphery of our freezing lesions, NF stains suggest the presence of increased neurofilament-containing structures, often in elaborate sprouts similar to those seen in the neocortical neuronal ectopias (cerebrocortical microdysgenesis) in layer I of the immune-disordered mouse (22). The overall appearance of lesions stained for NF that the increased staining on the periphery reflects a exuberant reaction to the lack of development of, or the injury to, neurofilamentous structures in the center of the lesion. The excess of NF-containing structures on the periphery of P0,P1 lesions could result from either increased sprouting of axodendritic processes or from the preservation of processes that would normally be eliminated. There is considerable evidence in the literature to support the latter argument. Nerve growth factor and other trophic factors are released during injury to nervous tissue (32–34). The presence of such trophic factors in the area of a lesion may diminish ontogenetic axonal pruning (35,36) as well as increasing sprouting.
Stains of glutamatergic processes also revealed widespread organizational abnormalities. Glutamatergic processes were markedly reduced in zones of neuronal loss and adjacent neuronal areas. Since glutamate is the principal neurotransmitter synthesized by cortical pyramidal neurons, and pyramidal neurons are specifically sensitive to hypoxic-ischemic insults (24), this finding was expected. The pronounced susceptibility of the latter neurons appears to be related to the neurotoxic effects of intracellular calcium, released during ischemia by the action of glutamate on NMDA receptors (24,37). What was not anticipated was that areas of layers V and VIa immediately adjacent to the necrotic zones, again apparently intact as viewed in Nissl stains, would show extensive distortion of glutamatergic processes. This may indicate that the ischemia-related cellular damage to susceptible neurons extends beyond the limits disclosed by routine cell stains.
In comparison with glutamatergic neurons, VIP cortical neuronal bodies in the area of the freezing lesion appear to have been remarkably spared, although occasional disturbances of their processes were noted. This discrepancy may be explained by the relative resistance of VIP interneurons to hypoxic-ischemic insults, in comparison with glutamatergic (pyramidal) neurons (23). GABAergic neurons are also resistant to hypoxic-ischemic insults (38) and may well be spared in our neocortical freezing lesions.
In conclusion, early focal injury to the cerebral cortex before completion of the period of neuronal migration, which we have imitated in the present study, may result in neuronal loss, laminar reorganization with the production of microsulcus (sulci), and minor secondary disturbances in neuronal migration. Immunocytochemical staining reveals that the injury affects anoxia/ischemia sensitive elements, the local connectional architecture as disclosed by neurofilament staining is disrupted, and structures at sites removed from the region of injury may be affected as well.
ACKNOWLEDGEMENTS
Supported by: NIH Grant HD 20806, grants from the Carl W. Herzog Foundation, the Milton Fund, and the Research Division of the Orton Dyslexia Society. Supported, in part (PH), by the University of Ottawa Department of Pediatrics Research and Development Fund.
The authors thank Antis Zalkalns for technical assistance, Steven Kleshinski for his help in fabricating the freezing apparatus, and to the reviewers for their helpful comments on an earlier version of the manuscript.