Note to the reader: This is a revised edition of a
paper published in Journal of Neuropathology and Experimental Neurology (1992;51:601–611).992;51:601-11
The definitive original print version is copyright ©2000 by The American Association of Neuropathologists.
New figures, text, and links have been
incorporated into the revision. Revised HTML (http://nervenet.org/netpapers/Rosen/MGDev92/MGDev.html)
copyright ©2001 by Glenn
D. Rosen
THE DEVELOPMENT OF INDUCED CEREBROCORTICAL MICROGYRIA IN THE RAT
Glenn D. Rosen, Douglas M. Press, Gordon F. Sherman, and Albert.M. Galaburda
Dyslexia Research Laboratory and Charles A. Dana Research Institute, Department of Neurology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA.
Address correspondence to:
Glenn D. Rosen
Department of Neurology
Beth Israel Deaconess Medical Center
330 Brookline Ave.
Boston, MA 02215
EMAIL: grosen@caregroup.harvard.edu
Placement of a freezing probe on the skull of neonatal rats produces four-layered microgyria, complete with a lamina dissecans and microsulcus. We studied the developmental course of this induced microgyria under light microscopy by examining changes in neurons, glia, and macrophages following a focal freezing insult on the day of birth (P0). The destruction of neurons and glia induced by the freezing probe extends through the cortical plate and occasionally through the subplate, but the pial membrane appears undamaged and radial glial cells, while damaged, are not eliminated. Reactive astrocytes and macrophages arrive in the damaged area within 24 hours of the injury, and repair of the damaged tissue peaks within the first week. Damaged radial glial fibers regrow, and supragranular neurons migrate through this damaged area, also within the first week. The newly formed supragranular layer overlies the cell-free area. The damaged cortex begins to assume its adult-like microgyric appearance from P5 to P10. On P15 and P32, long glial fibers, resembling radial glia, are present and are immunoreactive for GFAP and radial glial fiber antibodies (vimentin and Rat-401). No such fibers appear at this age in the non-microgyric areas or in normal brains. We conclude that microgyria formation may be the consequence of brain repair mechanisms occurring during neuronal migration to the neocortex, and that it appears to preserve primitive features characteristic of the developing cortex.
Four-layered microgyria (polymicrogyria) has been observed in association with several human developmental brain disorders (1–8). The origin of microgyria is not understood, but injury to the developing brain is widely accepted as a key pathogenetic factor (7,9–11). The timing of the injury, i.e., whether it occurs during and/or after completion of neuronal migration to the neocortex, is debated (7,10–12) , but the experimental evidence supports the hypothesis that the malformation originates during neuronal migration (13–16). Specifically, focal microgyria can be produced by applying a freezing probe to the skulls of newborn rat pups during, but not after, the period of neuronal migration to the neocortex, which continues for two or three days after birth in this species. The neuropathologic lesion resulting from this type of early injury, most likely caused by vascular, ischemic damage, is indistinguishable from the four-layered microgyria described in human neuropathology. The first layer of the microgyric cortex (referred to here as layer i) infolds and fuses to produce a microsulcus; layer ii, which is continuous with adjacent, normal layers II and III sweeps around the microsulcus; layer iii is devoid of cells and comprises the lamina dissecans; and layer iv, a thin strip of neurons, is continuous with normal layer VIb in uninvolved adjacent cortex (see Fig. 1). In contrast, injury occurring after the completion of neuronal migration results in gliotic, post-traumatic scarring (13,14,17).
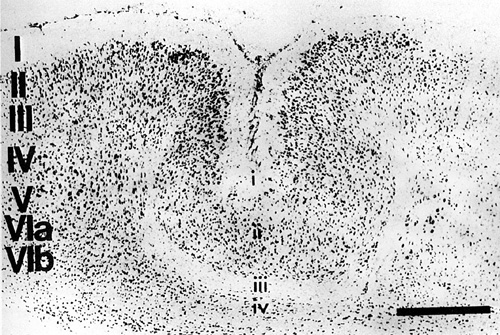 |
| Fig. 1 - Nissl-stained section of the cerebral cortex of an adult rat illustrating the induction of four-layered microgyria by a freezing insult at P0. Large Roman numerals denote laminæ of normal cortex, small Roman numerals denote the four laminæ of the microgyric cortex. Bar = 500 µm. |
The steps leading to scar formation after focal injury to the adult cortex have been discussed in detail (18–24) , and recent reports described the course taken by the developing cortex after freezing injury (16,25). Suzuki and Choi (16) placed a freezing probe on the skull of a neonatal (postnatal day 2) rat and followed the chronological sequence of neocortical repair. In the present paper, we seek to replicate and extend these findings. We employed classical cell stains to examine sequential changes in the architecture of the neurons, and immunohistochemical techniques to disclose developmental alterations in radial glia (Rat-401 and vimentin), astrocytes (GFAP and vimentin), macrophages (ED-1), and pial membranes (laminin).
Protocol
We obtained time-mated female Wistar rats (Charles River Laboratories, Wilmington, MA) on days 16–18 of gestation. Focal necrotic lesions were induced in the cerebral cortex of the pups from these mothers using a modification of the technique employed by Dvorák and colleagues (13,14) and described in detail elsewhere (15). On the day of birth (P0) pups were anesthetized (hypothermia) and a small incision was made in the antero-posterior plane of the skin over the left cerebral hemisphere, exposing the skull. A stainless steel probe (2 mm diameter at the tip) contained within a 50 ml centrifuge tube filled with methyl butane was cooled to -70°C with dry ice and then applied to the skull approximately midway between bregma and lambda, for 5 seconds. The skin was then quickly sutured, the animal warmed under a lamp, and returned to the mother.
Fifty-eight animals were sacrificed on either P0 (within 2 hours of the freezing insult; n=2), P1 (n=8), P3 (n=6), P5 (n=9), P7 (n=9), P9 (n=4), P10 (n=5), P15 (n=8), P17 (n=2), P21 (n=2), or P32 (n=3) under deep anesthesia (hypothermia or Pentobarbital depending on the age of the animal) by intracardiac perfusion of 0.9% saline followed by 2% paraformaldehyde/0.05% glutaraldehyde or 4% paraformaldehyde (both in 0.1 M phosphate buffer). Following perfusion, the brains were removed from the skulls, postfixed for 24 hr in the fixative, placed in a 10% sucrose solution in 0.1 M sodium phosphate buffer for at least 24 hr, and then placed in 30% sucrose buffer until the brains sank. A nick was made on the ventral surface of the right hemisphere, and the brains were frozen on dry ice, serially sectioned in the coronal plane at 30 µm, and stored in 0.1 M sodium phosphate buffer. One series of every tenth section was stained with Thionin for Nissl substance. Adjacent series were stained for glial fibrillary acidic protein (GFAP), radial glial fibers (Rat-401, Vimentin), macrophages (ED-1), and laminin.
Sections were examined under light microscopy
Histological Preparation
For all antibodies, a control series was run with the omission of the primary antibody and negative staining was seen.
Nissl
Mounted sections were stained for Nissl substance with 0.05% Thionin using standard techniques.
Glial Fibrillary Acidic Protein
Free-floating sections were rinsed twice in phosphate-buffered saline (PBS; pH 7.4) for five min each and transferred to a buffered 0.6% hydrogen peroxide solution to block staining of endogenous peroxidases. Sections were then placed in vehicle only for 20 min at room temperature. Sections were placed into a 1/25 solution of primary antibody (Incstar, Stillwater, MN) overnight at 4°C. The vehicle (diluent) for all antibody incubations was 5% goat serum in PBS. The next day, sections were transferred into a biotinylated goat anti-rabbit immunoglobulin solution (Vector Laboratories, Burlingame, CA) diluted 1/60 for two hr at room temperature. After two washes in PBS, the sections were placed into ABC complex (Vector Laboratories) for two hr at room temperature. The tissue was rinsed twice in PBS and twice in 50 mM Tris buffer (pH 7.6) and developed, dehydrated, counterstained, and coverslipped.
Radial glial fibers
Rat-401. This monoclonal antibody is directed against radial glial fibers and was provided by S. Hockfield, Yale University. Free-floating sections were rinsed twice in phosphate-buffered saline (PBS; pH 7.4) for five min each and transferred to a buffered 0.6% hydrogen peroxide solution to block staining of endogenous peroxidases. The sections were rinsed twice in PBS and incubated overnight at 4°C in a 1/4 dilution of the primary antibody. The diluent for all antibody incubations was 3% rabbit serum in PBS.
Sections were then placed into a solution containing the linking antibody (rabbit anti-mouse immunoglobulin - Dakopatts (Santa Barbara, CA) Z259 - diluted 1/20) at room temperature for two hr. The sections were rinsed twice with PBS and placed in a 1/250 dilution of mouse peroxidase anti-peroxidase (Dakopatts B650) at room temperature for two hr. The tissue was rinsed twice in PBS and then twice in 50 mM Tris buffer (pH 7.6) and developed using 0.05% diaminobenzidine and 0.005% hydrogen peroxide diluted in Tris. After rinsing with Tris, sections were mounted, dehydrated, counterstained and coverslipped.
Vimentin. This monoclonal antibody (Boehringer Mannheim, Indianapolis, IN) stains radial glial fibers and is somewhat less specific than Rat-401, staining astroglial filaments late during development. The protocol is the same as that for Rat-401 with the exception of the dilution (1/500) of the primary antibody.
ED-1. This antibody (Bioproducts for Science, Indianapolis, IN) stains macrophages and the procedure is the same as that for GFAP. The dilution is 1/2500.
Laminin. The procedure for laminin staining is identical to that for GFAP, with the exception of the dilution of the primary antibody (Chemicon, Temecula, CA), which is 1/750
P0 - P5
Evidence of tissue necrosis is clearly visible by 1.5 hours after the freezing insult, although effects of the freezing injury render tissue histology suboptimal. There is widespread death of neurons and damage of radial glial fibers. Although there is a breach of the external glial limiting membrane, the pial membrane is intact, as can be discerned in the laminin-stained sections (Fig. 2a,b). There is no evidence of reactive astrogliosis as this time.
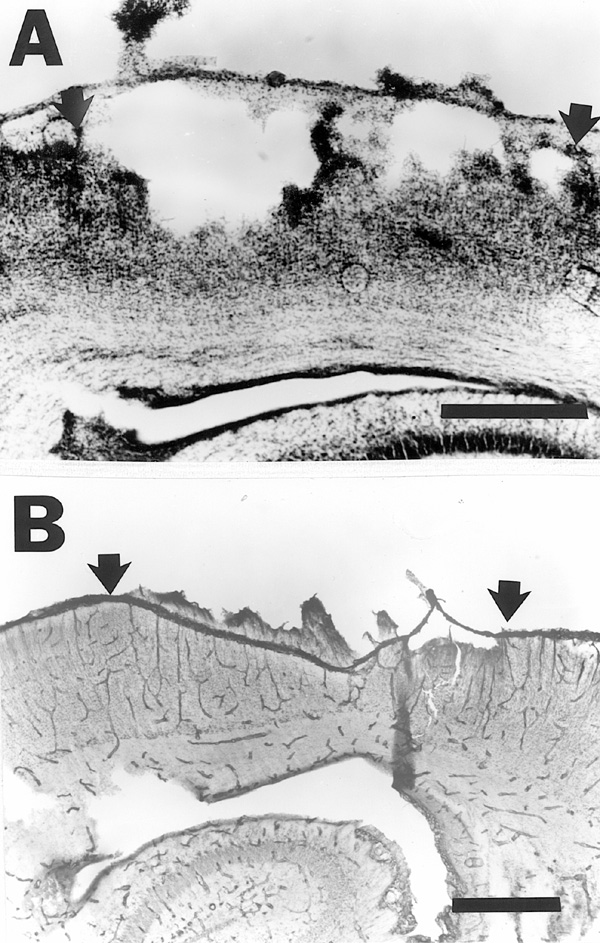 |
Fig. 2 - A. Nissl-stained section of the cerebral cortex of a P0 rat sacrificed 2 hours after the placement of a freezing probe on the skull overlying the area. Large arrows delineate the extent of damage. Note intact pial membrane. B. Section from the same animal as A immunohistochemically stained for laminin. Large arrows delineate the extent of damage. The pial membrane, although distorted by the mounting process, is intact. Bars for both panels = 500 µm |
Twenty-four hours after the freezing insult there is complete necrosis of the cortical plate. Occasionally the damage extends through the subplate to the white matter, and when this occurs, it usually underlies the center (the region underlying the center of the freezing probe) of the lesion. Most often, however, the subplate neurons (layer VIb) are spared (Fig. 3a). Reactive astrogliosis is seen (Fig. 3b), and there is some evidence of macrophage activity at this time (Fig. 3b,d). Radial glial fibers are still damaged in the area of tissue necrosis, but have not completely degenerated, and appear to be truncated (Fig. 3c).
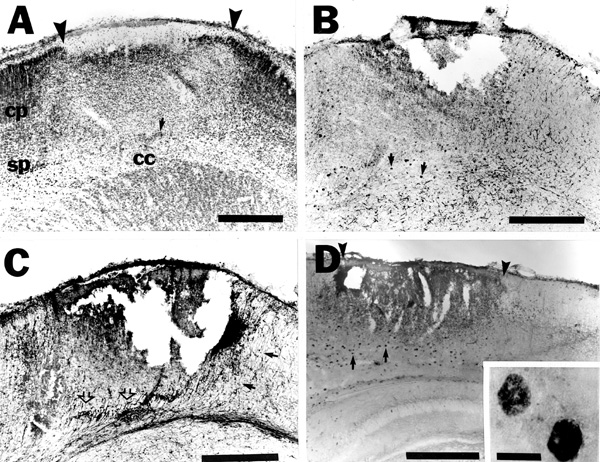 |
| Fig. 3 - A. Nissl-stained section of the cerebral cortex of a rat sacrificed 24 hours after placement of a freezing probe on the skull overlying the area at P0. Tissue necrosis (arrowheads) encompasses the cortical plate (cp), but spares much of the subplate (sp; small arrow denotes a subplate neuron) and the corpus callosum (cc). B. Section adjacent to A immunohistochemically stained for GFAP demonstrates reactive astrogliosis and macrophage activity (arrows; see panel D and Fig. 4c for more detail). C. Section adjacent to B immunohistochemically stained for radial glial fibers with Rat-401. There is a disruption of radial glial processes in the area of damage (open arrows), although intact fibers can be seen medial (arrows) and lateral to the damage. D. Cerebral cortex of a rat sacrificed 24 hours after placement of a freezing probe on the skull overlying the area at P0 immunohistochemically stained for macrophages with ED-1 (arrows and inset). This photomicrograph is from a different animal than that pictured in panels A-C. Bars for panels A-D = 500 µm. Bar for inset to panel D = 25µm. |
By P3 neurons are seen surrounding the area of necrosis (Fig. 4a), and radial glial fibers appear at the base of the lesion (Fig. 4b). The general appearance of the injured area at this time is “bowl-shaped” with repair proceeding at the base and the lateral edges of the damage. There are large numbers of GFAP-positive immunoreactive cells throughout the base of the lesion, most of which are astrocytes and some of which are reactive macrophages (gitter cells) as confirmed by ED-1 immunostaining. Under higher magnification, the GFAP-immunoreactive components of these gitter cells can be seen to be phagocytosed particles of GFAP-positive astrocytes (Fig. 4c).
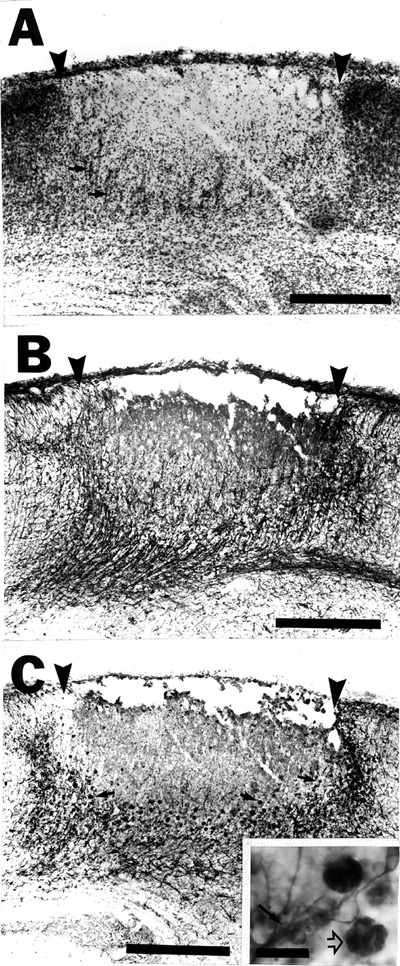 |
Fig. 4 - A. Nissl-stained section of the cerebral cortex of a rat sacrificed at P3 (72 hours after placement of a freezing probe on the skull overlying the area). Tissue necrosis (arrowheads) is somewhat more delimited than seen 48 hours earlier (Fig. 3a). B. Section adjacent to A immunohistochemically stained for radial glial fibers with Rat-401. C. Section adjacent to B immunohistochemically stained for GFAP demonstrating reactive astrogliosis (filled arrow in inset) and macrophage activity (arrows and open arrow in inset). GFAP-positive staining for macrophages (gitter cells) may be the result of immunoreactive phagocytosed astrocytes (open arrow in inset). Bars for panels A-C = 500 µm. Bar for inset to panel C = 25 µm. |
Incipient microsulci are evident at P5 and the general Nissl appearance of the lesion approximates that of the adult state. Some radial glial fibers can be seen crossing through the damaged area. Macrophage activity is present and reactive astrocytes remain in the area of lesion. Here, as is also the case on P3, there is no increase in astrocytic activity in the opposite hemisphere, as might have been expected (19,24).
P7 - P10
One week following the freezing insult, the lesion continues to resolve toward its mature appearance. One microsulcus or several microsulci are nearly fully formed along the anteroposterior axis of individual animals, and the neurons that make up layer ii of the microgyrus can be seen in their final locations. The cell free lamina dissecans is easily seen and is filled with reactive astrocytes and macrophages (Fig. 5a).
Rat-401-, vimentin-, and GFAP-immunoreactive fibers that have the general appearance of radial glia can be seen throughout the lesion. While it is not unusual for radial glial fibers to be immunoreactive to both GFAP and radial glial fiber antibodies (Rat-401, vimentin) during this stage of development in the normal rat (26–28), the number of GFAP-labeled fibers was much greater in and around the lesion than anywhere else. Rat-401 and vimentin immunohistochemistry revealed swollen distal fibers with opaque protuberances at the base and in other more superficial areas of the lesion (Fig. 5b,c). These processes have some characteristics of radial glial growth cones (Fig 5d; see ref 29) , and do not resemble the more triangular shape (and more distal location) of glial endfeet.
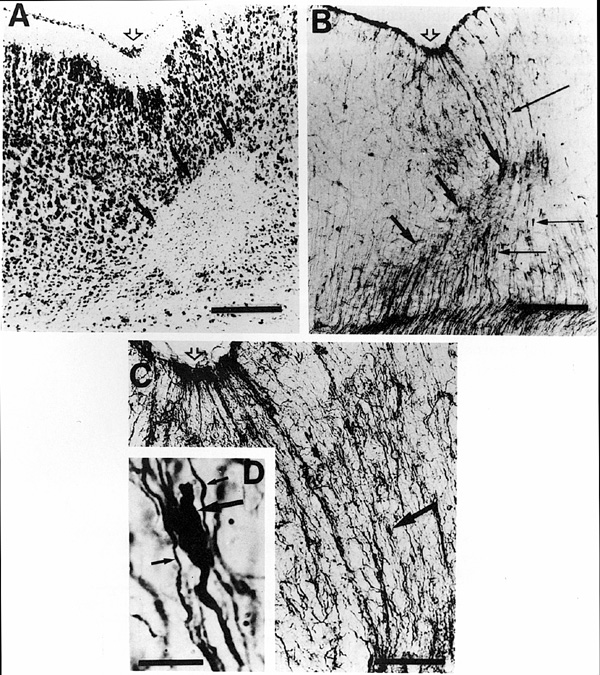 |
Fig. 5 - A. Nissl-stained section of the cerebral cortex of a rat sacrificed at P7 after a freezing insult at P0. This is taken from the periphery of the lesion (underlying the outer radius of the freezing probe) and illustrates the beginning of the microsulcus (open arrow) and the lamina dissecans (arrows). B. Section adjacent to A immunohistochemically stained for Rat-401 demonstrating radial glial-like immunoreactivity. Note enlarged protuberances at the ends of some fibers (long arrows) contained within the lamina dissecans (arrows) and dorsal to it (see C for greater detail). Open arrow for orientation with A. C. Enlarged portion of B showing immunohistochemically stained radial glial fibers (Rat-401) which can be seen spanning from the lamina dissecans to the pial surface. Open arrows indicate pial surface and aid in orientation with A and B. Small arrows denote "tadpole-like" structures, shown in higher power in D. D. Enlarged area from C demonstrating the "tadpole-like" structure at the end of a radial glial fiber. Bars for panels A, B = 250 µm. Bar for panel C = 100 µm. Bar for panel D = 10µm. |
The lesion, for the most part, has taken on its fully mature appearance by P10. Microsulci are present throughout the anteroposterior axis and the typical four-layered microgyria can be seen. GFAP immunoreactive fibers that resemble radial glial fibers in configuration and orientation are seen, although less frequently than in the P7 animal. Rat-401 immunoreactivity is nearly absent from the brain with the exception of the area of the lesion, where radial glial-like, long immunoreactive fibers are stained. These fibers can be seen at the base of the lesion as well as reaching down from the pial surface (see Fig. 7). Both radial glial fibers and astrocytes are immunoreactive for vimentin at this age. As with Rat-401, vimentin-positive radial glial fibers appear only in the area of the microgyrus.
P15 - P32
The microgyrus is fully formed. There is evidence of glial scarring along the base of the lesion and occasionally surrounding the microsulcus, as indicated by GFAP-positive staining (Fig. 6a,b). Rat-401- and vimentin-immunoreactive fibers, which are morphologically similar to radial glial fibers, are seen only in the area of damage (Fig. 6c,f) and can be seen to reach from the pial surface to the white matter (Fig 7). GFAP-positive astrocytes continue to be present in their expected locations (i.e., in the lamina dissecans), and some fibers that resemble the long filamentous morphology of radial glial fibers show GFAP-like immunoreactivity (Fig. 6e). Macrophages, while still present, are substantially diminished in numbers.
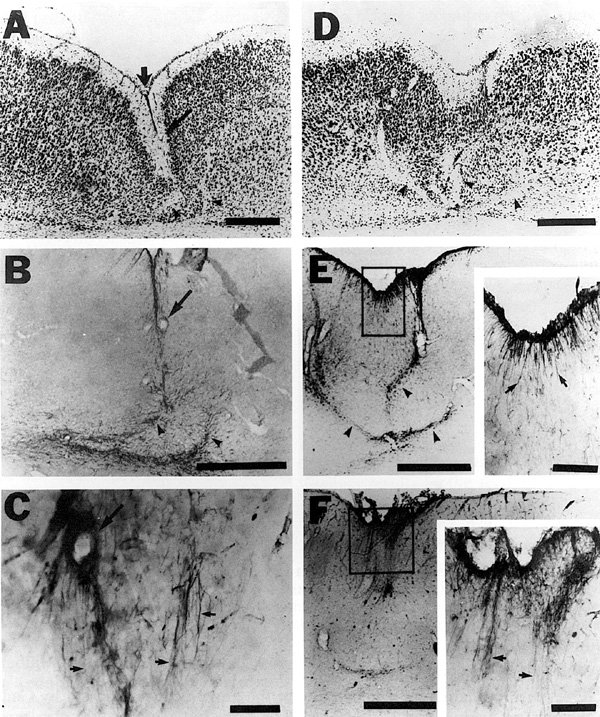 |
| Fig. 6 - A. Nissl-stained section of the cerebral cortex of a rat sacrificed at P15 after a freezing insult at P0 demonstrating typical area of induced microgyria complete with lamina dissecans and microsulcus (broad, vertical arrow). Diagonal arrow denotes blood vessel for orientation with B and C. Bar = 500 µm. B. Section adjacent to A immunohistochemically stained for GFAP illustrating reactive astrogliosis (arrowheads) in the area of the lamina dissecans. Bar = 500 µm. C. Section adjacent to B immunohistochemically stained for radial glial fibers with Rat-401. Radial glial fiber-like immunoreactivity is only seen in the area of the microgyria (arrows). Bar = 50 µm. D. Nissl-stained section of the cerebral cortex of a rat sacrificed at P21 after a freezing insult at P0 showing an induced microgyria with lamina dissecans (arrowheads). Bar =500 µm. E. Section adjacent to D immunohistochemically stained for GFAP demonstrating reactive astrogliosis (arrowheads) in the area of the lamina dissecans. Bar = 500 µm. Inset is enlargement of GFAP-positive staining for glial filaments that resemble radial glial fibers (arrows; Bar = 100 µm). F. Section adjacent to E immunohistochemically stained for radial glial fibers with Rat-401. Bar = 500 µm. Inset is enlargement of Rat-401-positive staining for radial glial fibers (arrows; Bar = 100 µm). |
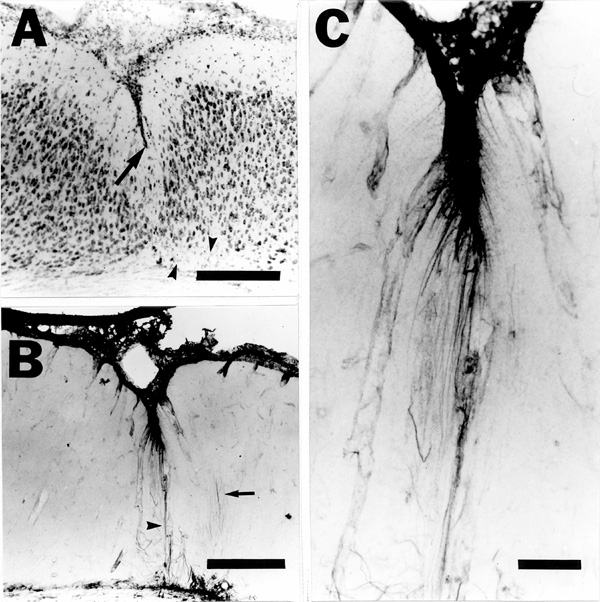 |
Fig 7 - A. Nissl-stained section of the cerebral cortex of a rat sacrificed at P32 after a freezing insult at P0 showing induced microgyria complete with lamina dissecans (arrowheads). B. Section adjacent to A immunohistochemically stained for vimentin demonstrating radial glial-like fibers in the area of damage reaching from pial surface to white matter (arrowhead). Note also immunoreactive fibers adjacent to the area of damage (arrow). Bar for A and B = 250 µm. C. Higher power photomicrograph of section B. Bar = 50 µm |
Acute changes to the neocortex following injury
The immediate effects of the freezing insult
The freezing insult causes complete tissue necrosis of the area underlying the freezing probe, as was reported previously (16,25). Neurons in the cortical plate (presumptively layers IV-VI), GFAP-immunoreactive cells, and radial glial fibers are either destroyed or disrupted. The effects of the injury appear to be uniform and severe in the area underlying the center of the probe, with complete destruction of the cortical plate (and occasionally the subplate), and somewhat less severe at the lateral edges. Although radial glial processes are initially damaged by the freezing insult, the cell bodies themselves are not destroyed, most likely because they are located close to the ventricle, beyond the reach of the freezing injury (12).
Despite the severe nature of the freezing insult, the pial surface appears intact as early as 1.5 hours after application of the freezing probe. The external glial limiting membrane that lies directly beneath it, however, is destroyed. This indicates that either the freezing insult does not disrupt the pial membrane, or the latter is quickly repaired after damage. We had hypothesized earlier (15) that the freezing insult produces ischemia. If that is the case, the pial membrane may be spared because it is protected by cerebrospinal fluid, collateral vascularization, and/or lower metabolic requirements.
Local reaction to injury
The astrocytic reaction to the freezing insult is quick, with the first increase of astrocytes in the base of the area of the damage occurring within 24 hours, and reaching their highest concentration 48-96 hours later. By P3, a large number of macrophages are seen in the areas surrounding the lesion, as was reported previously (16). Both these astrocytes and macrophages are restricted in their distribution mostly to what will eventually be layer iii of the microgyrus—the lamina dissecans—by P5. The arrival of reactive astrocytes within the first 24 hours following the injury agrees with the report of Janeczko (30) who, using [3H]thymidine-labeling, showed reactive astrocytic proliferation within 24-48 hours following injury to the neonatal cortex. In that study, brains were not examined until P40, so it is not known exactly when these proliferating astrocytes appeared in the damaged area. A similar time frame is also seen following freezing insults to the cerebral cortex of adult rats (19,20) ,
In contrast to both Anders et al. and our findings, Amaducci et al. (19) found an increase in immunoreactive astrocytes in the hemisphere opposite the lesion within 24 hours of injury, a discrepancy perhaps explained by the greater severity of the freezing insult (5 mm probe cooled with liquid nitrogen for 30 sec). In support of this explanation, extensive laser-induced lesions of the adult rat neocortex also result in distal astrocytic reactions (24). At the other extreme, Berry et al (31) found no astrocytic reaction after neonatal stabbing lesions of the cerebral cortex. Possibly the extent of injury affects the appearance of astrocytic proliferation and disposition.
An important factor in reestablishing the integrity of the damaged brain is the repair of the external glial limiting membrane. In the case of freezing injury to the adult brain, this repair occurs 1-2 weeks following the initial insult (32). Without electron microscopy, we are unable to determine with certainty whether the external glial limiting membrane is repaired as others have noted (16) , however glial endfeet reappear subjacent to the pial membrane, and an increase in GFAP immunoreactivity can be seen in this region by P7. The morphology of these GFAP-immunoreactive cells resemble that of radial glial fibers more than that of mature fibrillary astrocytes seen in other regions of the brain, and this morphological distinctiveness remains unchanged at least until P32. Rat-401-immunoreactive fibers can be seen in the same locations and have a similar morphology; we have not performed double-labeling experiments to determine whether these fibers express both antigens.
Chronic changes to the neocortex following injury
The longevity of radial glial fiber-like immunoreactivity
During the first postnatal week of normal rat cortical development, radial glial fibers are immunoreactive for vimentin and Rat-401 only. Starting during the second week of life, the fibers stain positively for both GFAP and radial glial antibodies. By the third week of life, vimentin- and GFAP-positive staining is seen in mature astrocytes and there is no evidence of radial glial fibers by immunohistochemical or morphological criteria. Thus, it is likely that radial glial cells transform to astrocytes starting around the first week after birth, with disappearance of radial glial fibers by the third week (26,33).
This developmental time course is supported by our findings. In the undamaged hemisphere, Rat-401-positive fibers, morphologically identifiable as radial glial cells, are seen from P0 to P10. By P15, there are few Rat-401-labeled fibers and none are seen on P17 or beyond. These findings were confirmed with vimentin. A few radial glial fibers stain positively for GFAP on P0, and their numbers increase until P7. By P10, there only a few GFAP-positive stained fibers that have radial glial morphology, and there is no remaining staining of this type by P15.
A different picture emerges in the damaged area, however. Beginning around P3, there appears to be a regrowth of radial glial fibers through the area of damage. By P7, the “tadpole-like” endings, similar in external appearance to the growth cones reported by Gadisseux et al. (29) in E14 mice, but which could also represent pathological processes, can be seen in both Rat 401- and vimentin-positive cells. Examination of their fine structure is required for definitive classification as growth cones or injured endfeet. In either case, release of neurotrophic factors by the reactive astrocytes (34-36) may be responsible for radial glial regrowth through the damaged area, which provides a skeleton for the resumption of neuronal migration to the upper layers.
In the injury case, GFAP and Rat-401 immunoreactive fibers resemble radial glial fibers until at least P32, but it is not known whether they represent normal radial glia. It could be that these fibers are neither typical astrocytes nor radial glial cells but some hybrid cell peculiar to this type of damage. Alternatively, it is possible that the normal transformation of radial glia to mature astrocytes is prevented, resulting in a cell that has both features of radial glia and astrocytes. Thus the glial cell maintains its original morphology and antigenicity. Finally, there could be an increase in proliferation of radial glial cells, as well as astrocytes, as a result of the freezing damage. Thus, those radial glial cells that were present during the time of freezing injury hasten their transformation to reactive astrocytes as a reaction to the cortical damage. This in turn stimulates the generation of new radial glial cells through the release of trophic factors (34-36). These newer radial glial cells may have a longer lifespan than earlier generated cells and may or may not transform to astrocytes by P32. Currently, we cannot distinguish among these possibilities.
Formation of the microsulcus
The mechanism for cortical folding in the normal brain, while not known, has engendered much speculation and modeling (37-45) , and has been influenced by the study of microgyria (3,46,47). Can the formation of a microsulcus following a freezing insult to the developing rat brain be explained by any of these hypothesized models? Richman et al. (47) considered that cortical folding is caused by unequal forces generating differential growth of the outer cortical laminæ (layers I–III) vs. the inner cortical laminæ (layers IV–VI). Because their mechanical model was based in part on the empirical findings of human microgyria, rather than normal cortical folding, it is perhaps not surprising that it also fits with our data. Specifically, the freezing injury destroys the cortical plate, which at the time of the injury contains only those cells of the inner cortical laminæ. With the subsequent migration of upper laminæ cortical neurons, the stresses in the upper cortical laminæ are much greater than that of the lower cortical laminæ and hence the cortex buckles.
Smart and McSherry (43) correctly point out that Richman et al.’s mechanistic approach does not take into account the inherent radial structure of the developing neocortex. This is less of a concern in our case, in that freezing insult completely disrupts any inherent focal radial structure in this system. On the other hand, radial glial fibers remain intact in the areas immediately surrounding the freezing-induced damage, suggesting that these radial structures may play a role in the eventual dramatic reorganization of the neocortex.
An alternative hypothesis proposed that the cortex will tend to buckle where the it is thinnest (40). This hypothesis could also explain the formation of the microsulcus following neonatal freezing lesions. In this case, the structure of the cortex is severely compromised by the freezing injury. As the brain matures and grows, the damaged area is thinner compared to the normally developing cortex on either side of the damage. The adjacent cortex will then collapse into the thinner, damaged area.
Lissencephaly vs. polymicrogyria
It is interesting to note the differences between the four-layered cortex associated with genetic defects leading to lissencephaly (e.g., the Miller-Dieker malformation), and the present form of microgyria. While the first and third layers are similar in the two malformations, the former has a thinner second layer, with large cells, and a much thicker fourth layer. This fourth layer in the lissencephalic cortex merges with an underlying mass of gray matter present in white matter. The generally accepted explanation for the formation of lissencephaly is that there is an arrest of the second wave of neuronal migration such that neurons destined for the supragranular layers cannot migrate past the previously arrived subgranular cells (37,48). The second layer, with its large cells, therefore represents early migrating pyramidal neurons ordinarily destined to populate layers V and VI. Conversely, in the present form of microgyria, layer ii neurons are smaller and most likely represent supergranular neurons. On the other hand, most layer V neurons are destroyed and non-existent in the present malformation. This fundamental difference—an inherent disturbance of neuronal migration on the one hand and the brain’s reaction to damage on the other—may explain the obvious differences in the neuropathologic appearance of these two malformations.
Conclusions
We propose that the following temporal sequence of events result in the formation of focal microgyria following a freezing insult on P0:
The freezing insult induces complete or partial necrosis of all cells and processes directly underlying the probe: The external glial limiting membrane is damaged, but not the pia.
Reactive astrocytes arrive quickly in the damaged area and promote the repair of the external glial limiting membrane and, along with macrophages, phagocytose damaged tissue. In addition, these astrocytes may secrete neurotrophic and other growth factors that may influence steps (3) and (4).
The regrowth of radial glial cells through the damage aids in the continued migration of neurons through the area of damage.
Following repair of the external glial limiting membrane, long glial fibers that are Rat-401-, vimentin-, and GFAP-immunoreactive are seen well past the time they normally disappear.
The microsulcus is formed by the migration of neurons through the area of damage to the upper cortical layers and perhaps by the relative collapse of the cortex around into the damaged area.
Microgyri are not formed following freezing insults after the time of neuronal migration is completed (13,14,16,17) which suggests that the microgyria in this model is the consequence of brain repair mechanisms that include migration of neurons after the injury.
ACKNOWLEDGMENTS
This work was supported by: NIH Grant HD 20806, grants from the Carl W. Herzog Foundation, the Milton Fund, and the Research Division of the Orton Dyslexia Society. The authors thank Drs. Susan J. Hockfield and Daniel H. Geschwind for generously supplying the Rat-401 antibody, Drs. Miguel Marin-Padilla and Barbara L. Finlay for their helpful comments on an earlier version of the manuscript, and Lisa V. Stone, and Judy M. Richman for their technical assistance.