![]()
Note to the reader: This is a revised edition of a paper published in Time, Fluency, and Dysleixa, M. Wolf, editor York Press, Timmonium, Maryland, 2001. Copyright ©2001, National Dyslexia Research Foundation.
New figures, text, and links have been incorporated into the revision. Revised HTML (http://nervenet.org/netpapers/Rosen/CreteRev2001/CreteRev.html) copyright ©2001 held by Glenn D. Rosen.
ANIMAL MODELS OF DEVELOPMENTAL DYSLEXIA: IS THERE A LINK BETWEEN NEOCORTICAL MALFORMATIONS AND DEFECTS IN RAPID AUDITORY PROCESSING?
Glenn D. Rosen,1 R. Holly Fitch,2 Matthew G. Clark,3 J. J. Lo Turco,4 Gordon F. Sherman,5 and Albert M. Galaburda1.
1Dyslexia Research Laboratory and Charles A. Dana Research Institute, Behavioral Neurology Unit, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, Massachusetts; 2Program in Biobehavioral Science, University of Connecticut, Storrs, Connecticut; 3Center for Molecular and Behavioral Neuroscience, Rutgers University, Newark, New Jersey;4Department of Physiology and Neurobiology, University of Connecticut, Storrs, Connecticut; 5The Newgrange Educational Outreach Center, Princeton, New Jersey.
Correspondence to:
Glenn Rosen
Department of Neurology,
Beth Israel Deaconess Medical Center
330 Brookline Avenue
Boston, MA 02215
USA
Email: grosen@caregroup.harvard.edu
Work in our laboratory has focused on the investigation of the link between neocortical malformations and defects in rapid auditory processing—traits that are purported to co-exist in dyslexics. We have specifically concentrated on two different model systems: An induced malformation (microgyria) in otherwise normal rats, and a spontaneously occurring malformation (molecular layer ectopia) in immune-disordered mice. Male rats with microgyria have defects in the discrimination of rapidly presented auditory stimuli that can be seen in both operant and reflex modification tasks. Female rats with equivalent microgyria, on the other hand, discriminate rapidly presented auditory stimuli as well as shams. Measuring neuronal sizes in the medial geniculate nuclei of these animals demonstrated that among males, there were more small and fewer large neurons in the microgyric animals as compared to sham operated animals. In contrast, there was no such neuronal size difference in females. We have evidence to support the notion that some of the widespread effects of neocortical malformations are related to changes in connections between the microgyric cortex and thalamus and to the cortex of the opposite hemisphere. Specifically, we found absence of thalamocortical fibers in the malformation and a preponderance of heterotopic callosal connections.
There are defects in rapid auditory processing among mice with spontaneously occurring molecular layer ectopias as well. When presented with an auditory gap detection task, mice with ectopias were significantly worse than non-ectopic mice at detecting a 5-msec gap (the shortest gap detected overall), but were not impaired at longer gap durations. Differences between ectopic and non-ectopic mice can also be seen at the level of neurophysiology. Specifically, we recorded the auditory event related potentials (AERPs) associated with two paired stimuli presented at intervals of varying durations. The second of the two AERPs was attenuated in the ectopic mice only during short ISIs, again supporting the notion of a disruption in rapid auditory processing by ectopic mice.
In sum, converging evidence from two animal systems suggests that there is a link between neocortical malformations and defects in rapid auditory processing. These results suggest that focal disruption of neocortical development can have specific effects on neurophysiological, neurobehavioral, and neuroanatomic systems.
INTRODUCTION
On the surface, the notion that one could learn anything about language disorders from the study of animal models can, at the very least, be called into question. There are certainly many factors seemingly working against our understanding of these complex behaviors through the use of animal models. The existence of language in non-human higher primates is not universally accepted, and there is certainly little or no evidence of linguistic behavior in any other non-human species. Given this fact, it is difficult to imagine what an animal model of dyslexia or other language disorder might entail.
That being said, recent investigations into the anatomical, psychophysiological, and neurophysiological substrates underlying developmental dyslexia and other language disorders have uncovered a variety of distinct attributes that characterize this disorder. Importantly, these findings have provided the foundation for their further examination using non-human subjects. In this chapter, we will review evidence of some of the biological and behavioral substrates underlying developmental dyslexia, concentrating primarily on focal malformations of the neocortex and defects in that ability to process rapidly changing sensory information. We will then discuss animal models that have proven useful in our understanding how these two categories of deficits may be related.
Developmental Dyslexia - Behavioral Phenotype
The definition of developmental dyslexia has proved to be quite controversial since the disorder was first described (1–3). In practice, dyslexics are usually diagnosed by a discrepancy between their expected and actual reading level (4,5). This is not to say the developmental dyslexia is simply a disorder of reading, as there is much evidence to support the notion that dyslexics have difficulties with a number of other cognitive skills including phonological awareness (e.g., 6–12) and verbal working memory (e.g., 6,9,10,12–19), Not all the differences seen between dyslexics and controls favor those who acquire reading in a standard fashion. Dyslexics are not worse than controls in all cognitive tasks. For instance, dyslexics are significantly better than normal reading controls at some orthographic skills (11,20), and visuospatial measures, including the WISC Block Design (21–23).
Developmental Dyslexia - Anatomy and Physiology
From the earliest writings on the subject, researchers had believed that there was a neural substrate underlying developmental dyslexia (3), but it was not until relatively recently that researchers began to systematically examine this issue. Drake (24) reported a small corpus callosum as well as “excessive gyration” in the brain of a post-mortem dyslexic, but that work was not continued in greater depth. Galaburda and colleagues have since systematically studied the brains of post-mortem dyslexics, and have found three anatomical differences between the brains of dyslexics and non-dyslexics. These include (1) symmetry of the planum temporale, a language-related region of the neocortex, (2) the presence of focal malformations of the cerebral cortex, and (3) defects in primary sensory systems. Although symmetry is an important issue, and one that has been successfully modeled in animals (25–29), we will concern ourselves only with the latter two traits in this chapter as they more directly relate to the other chapters in the volume.
Minor Malformations of the Cerebral Cortex
Microscopic examination of human dyslexic brains has revealed several related forms of developmental neuropathologic malformations (30–32). Five consecutively-studied male dyslexics and three female dyslexics have foci of cerebrocortical microdysgenesis consisting of (1) neuronal ectopias in neocortical layer I, (2) subjacent laminar dysplasia, (3) focal microgyria, and (4) microvascular anomalies. These abnormalities range in number from 30 to 150 focal lesions per brain, tend to be located in perisylvian regions, affect the anterior vascular borderzone, and usually involve the left more than the right hemisphere (see Figure 1). In two of the female brains, ectopias were relatively uncommon. Instead, these brains have large numbers of focal, myelinated glial scars that are located in the same distribution as the ectopias (32). Such scarring represents the same pathogenetic mechanisms as that of the ectopias, but acts on the developing brain somewhat later, after completion of neuronal migration, when the brain is no longer able to react by producing ectopias or microgyria. By contrast, the types of pathology described above are substantially less frequent, and when present less severe, in normative brains from comparable studies (33). Together, the different types of developmental neuropathology implicate a developmental window beginning early during the second half of pregnancy and terminating by the end of the second year of postnatal life.
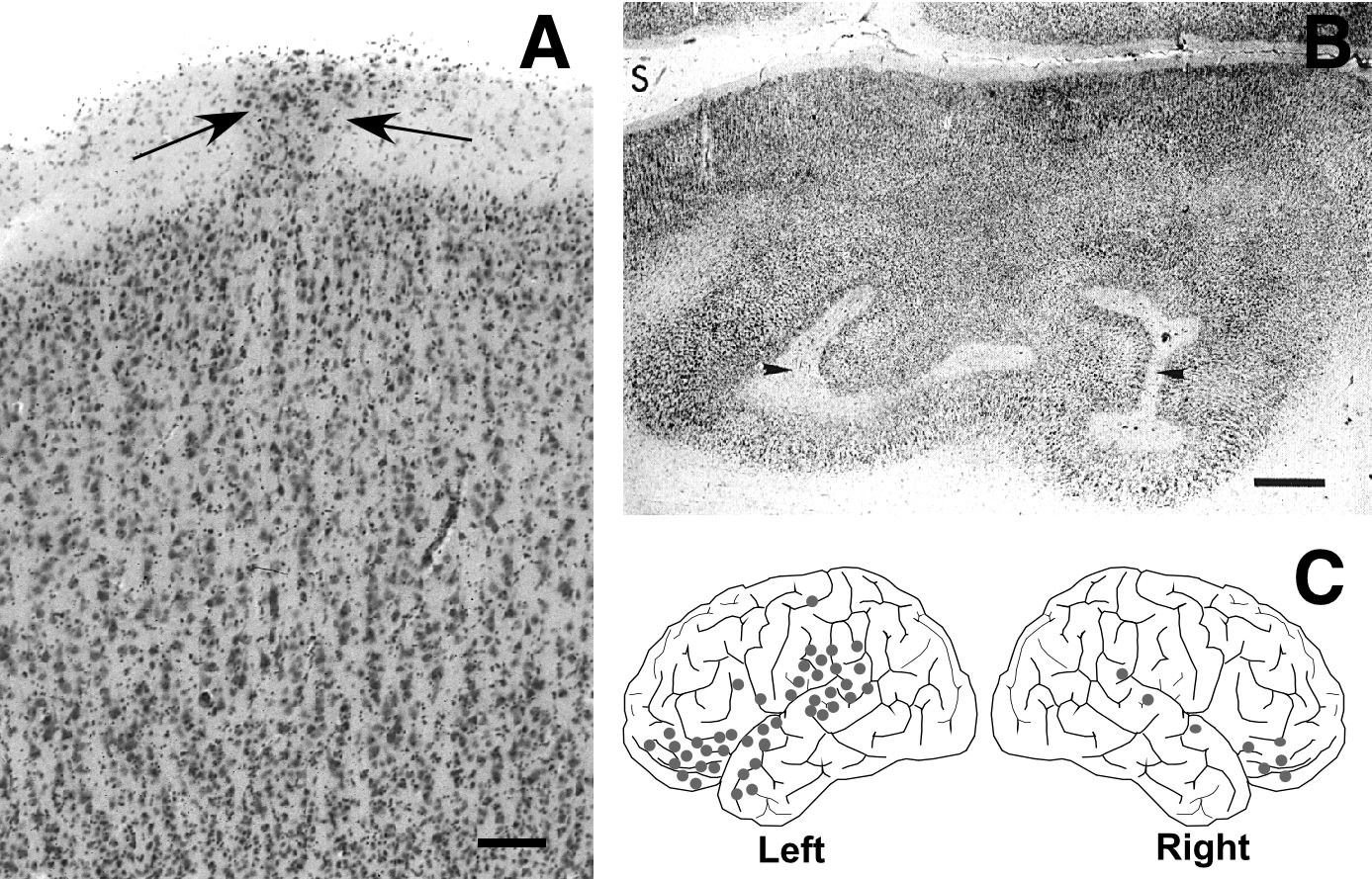 |
Figure 1 - Minor neocortical malformations in the dyslexic brain. A. Photomicrograph of molecular layer ectopia in layer one of a human dyslexic (arrows). Note the paucity of cells in the outermost layer of the adjacent cortex. Bar =400 µm. B. Region of microgyria in human dyslexic brain (arrowheads) Bar = 1 mm. C. Schematic demonstrating typical locations of molecular layer ectopias in the left and right hemispheres. Abbreviation: S = Sylvian Fissure. |
Multisensory Deficits in Fast Processing Systems
Developmentally language-impaired children (a large subset of whom are later diagnosed with dyslexia) suffer from rapid auditory processing deficits affecting even non-linguistic sounds (34–37). Researchers in the visual system (see Stein, Eden this volume 38–46) and somatosensory system (47) have shown similar deficits affecting temporal and spatial processing of stimuli in dyslexics, indicating that multiple sensory systems are involved. Livingstone and colleagues (43), for example, found that the magnocellular component of the visual system, which is responsible for processing fast, low-contrast information, is impaired in dyslexics. The physiologic response of this system is slowed in comparison to controls and in addition, the magnocellular neurons of the lateral geniculate nucleus (the primary thalamic visual nucleus) are smaller than in normals (43). Moreover, examination of neuronal size in the medial geniculate nucleus (the primary thalamic auditory nucleus; MGN) found more small and fewer large neurons in the left MGN, whereas there was no difference in neuronal size in controls (48). These findings complement previous reports of anomalies in the dyslexic MGN (49), and are consistent with reported behavioral findings of a left hemisphere-based phonological defect in dyslexic individuals (50,51).
Questions to be addressed
Dyslexia is a complex behavioral syndrome that includes at least disordered language processing, namely deficits in phonological awareness, and low-level, perceptual deficits, namely slowed visual and auditory temporal processing of low-level stimuli. One argument in the current scientific dialogue states that the cognitive problems of developmental dyslexia (both linguistic and non-linguistic) are the consequence of low-level sensory processing problems. Specifically, it is posited that language processing centers require the “correct type” of information from low level centers during development—without such information, language processing will be impaired (e.g., 52,53). An alternative hypothesis states that language and non-language sounds are processed separately even at low levels, and a problem in the latter would not necessarily translate into a problem with the former (e.g., 54). Further, it may be that low-level processing deficits are not the cause, but rather the consequence of high-level dysfunction. Namely, if high-level processing areas do not develop properly, they may not reinforce development of low-level processing areas for some functions, such as rapid auditory or visual processing, because they are incapable of processing those stimuli further. A third possibility, not negligible in developmental disorders, is that pathology is acquired at multiple levels at the same time. An example of the latter mechanism is a form of cerebral palsy in which circulatory deficiencies early in life lead to injury to the cortex as well as to subcortical gray masses (55,56).
In considering the two types of anatomic/physiologic deficits described above, one could, on the surface, provide support for any of the arguments above. The presence of cortical malformations, located primarily in perisylvian regions, could indicate that there are fundamental structural disturbances at the cortical level that may disrupt high-level processing. The disruption of cellular architecture at the thalamic level, on the other hand, could lend support to difficulties occurring at lower levels of processing. The nature of the human material limits further investigations along these lines, and we have therefore chosen to begin to tackle some of these issues through the use of rodent models. We have developed two models of minor malformations of the cortex—one spontaneous and one induced—that we have used to explore the anatomic, physiologic, and behavioral consequences of injury to the neocortex during development. These models have suggested a strong link between neocortical malformations and fast processing systems. In the next section of this chapter, we discuss the anatomic details of these models before turning to a discussion of the behavioral and physiological consequences of these malformations.
SPONTANEOUS AND INDUCED MINOR MALFORMATIONS OF THE CORTEX
Spontaneous malformations
Our animal model of spontaneous malformations was inspired by the reports of a link among dyslexia, left-handedness, and autoimmune disease (57–59); (but see also 60,61) . This link led us to examine the brains of immune-disordered mice where we found malformations similar in appearance to those seen in dyslexics. Specifically, in a series of studies, Sherman and colleagues (62–64) described molecular layer ectopias appearing in a number of strains of autoimmune mice, including the New Zealand Black (NZB) and BXSB. In addition to these “ectopias,” these mice also have cell-free, gliotic punched out lesions in the cortical plate (Figure 2A).
Overall about 40–60% of NZB and BXSB mice have ectopias. Typically, only one ectopic nest is seen in each affected brain, although up to 25% of the affected brains have multiple ectopias. Examination of the brains of fetal NZB mice showed that the earliest ectopias were present by embryonic day (E) 15 and, like the newborn (gestation is 19 days in the mouse), are associated with a disorganization of radial glial fibers and a breach (which is suspected to be caused by an injurious process) in the external glial limiting membrane (65). A subsequent study indicated that the small, focal breach in the external limiting membrane is created before E12 (the mechanism is unknown) and migrating neurons born during the period covering E12–18 migrate through this break (or are pushed through by later migrating neurons) leading to the formation of ectopias (66). The formation of ectopias is therefore secondary to events that occur before the end of neuronal migration in the mouse, a period corresponding to about the 4th or 5th month of gestation in the human.
In adulthood ectopias exhibit abnormal patterns of architecture. VIP, NPY, GABA and somatostatin containing neurons are seen in a small number of neurons within the ectopias in the NZB mouse (67). VIP and somatostatin are present in the largest numbers. Further, there was an increase in the total number of VIP neurons in the hemispheres with ectopias as opposed to those without ectopias. This difference was accounted for by more VIP neurons in the columns containing ectopias than in those in the homologous areas of the opposite hemispheres, as well as by more VIP neurons located medial to the ectopias. This indicated that the ectopias may represent not only inappropriately placed neurons, but also a problem with the regulation of their numbers. Subtle abnormalities are also seen in the hippocampus (68), and the cerebellum of the NZB was found to be unusual in a number of anatomical characteristics (69).
Several studies addressed the relationship between cortical anomalies and immune status, but none could be found. Specifically, embryos from BXSB mice were transplanted at the 8-cell stage into the uteri of control mice. There were significant effects on behavior and immune status, but there was no effect on the incidence of the malformations (70), suggesting the possibility of a genetic influence on the formation of the ectopias. Further research showed that ectopias are a recessive trait with incomplete penetrance and may be linked to multiple genes (71–73).
NZB mice with ectopias learn differently than those NZB mice without ectopias (74–76). Thus, ectopias depress performance of black-white discrimination in a swimming T-maze, increase the time necessary to find a hidden platform in the Morris maze (75), and interact with pawedness to affect performance on a spatial water escape task (74). Importantly, post-weaning enriched environment ameliorates the decrements seen in the discrimination learning task and the Morris maze. Thus, following a relatively short period of environmental enrichment, the behavior of animals with ectopias was indistinguishable from that of controls. This suggests the possibility that early experience can compensate for early brain injury (75,77).
Recent work by Denenberg and colleagues has pointed to some interesting parallels between the working memory deficits seen in dyslexics and those seen in mice with ectopias. Working memory requires an animal to disregard part of the information it has acquired on previous training trials and focus upon new information presented in the immediate trial. In a delayed-matching-to-sample task (DMTS) on a water maze, non-ectopic mice took less time and swam a shorter distance to find the platform on trial 2 than did ectopic mice (78). In a water version of the radial arm maze, ectopic BXSB mice made significantly more working memory errors (79).
There are also tasks where ectopic mice perform in a superior manner to their non-ectopic counterparts. In the Morris maze, for example, ectopic mice take less time to find a platform and are faster swimmers than non-ectopics (80). This group also studied mice that were embryo transferred to hybrid non-autoimmune mothers. Here, too, ectopic mice were better than non-ectopics on the Morris maze, suggesting that epigenetic maternal/uterine factors do not influence the behavioral consequences of these anomalies. Ectopic mice also have superior long-term retention of a water escape task (77).
Summary
A number of mouse strains that were bred to develop immune disorders have shown cerebrocortical malformations similar to those seen in dyslexia. These anomalies appear to be caused by injury during the early stages of cortical development. The etiology of this injury remains unknown, but it has been shown not to be related to the autoimmune disease itself, but rather to be under genetic control. These ectopias have both positive and negative behavioral effects, and some of the latter can be ameliorated by early experience.
In the following section, a complimentary animal model is introduced which involves the induction of cortical malformations.
Induced cerebrocortical malformations
In order to study further the effects of neocortical malformations, we wished to develop a model where the location and severity of the malformation could be controlled. Moreover, it was desirable to separate the other biological associations (e.g., immune-disorders) from the anatomy. Using a model developed originally by Dvorák and colleagues (81,82), we induce focal microgyria by placing a freezing probe (-70°C) on the skulls of newborn rats for approximately 5 seconds (83). The immediate effect of the freezing injury is to create a delimited region of cellular necrosis within 24 hours. Three days post-lesion, radial glial fibers regrow though a region of intense astrogliosis, and neurons begin to migrate through that area. The beginning of the formation of a microgyria can clearly be seen by day 5, starting primarily at the periphery of the lesion, where radial glial fibers are continuing their regrowth through the damaged area. The microgyria gain their adult appearance by P15 at which time radial glial fibers can still be seen in the area of damage, although not elsewhere (see Figure 2B). These results suggest that the formation of microgyria is the result of basic brain repair mechanisms occurring during the end of the period of neuronal migration (84,85).
Although occasional layer I ectopias arose following freezing injury, they were of a qualitatively different type from those seen in immune-disordered mice. The collections of neurons in ectopias always reach the pial surface and they generally have a contained, “mushroom-like” appearance, whereas the induced anomalies tend not to reach the pial surface and are more dispersed. Based on the finding of a breach in the external limiting membrane overlying areas of spontaneous ectopias (65), we created a small focal area of damage of the external limiting membrane by puncture wound of the neocortex. This resulted in ectopic collections of neurons in layer I of the neocortex comparable to those seen in spontaneous ectopias of humans and mice (86).
Nonimmune-disordered mice with induced ectopias and microgyria have behavioral deficits that are, in some ways, quite similar to those seen in immune-disordered mice with spontaneous ectopias. Lesioned mice performed poorly when compared to sham-operated animals in discrimination learning, in a spatial Match-to-Sample task, and in a Lashley Type III maze. In shuttlebox avoidance conditioning, where immunological disorder compromises behavioral performance, there was no difference between lesioned and sham animals. These results reveal the similarities to the behavioral effects of spontaneous and induced neocortical malformations (87).
Back to “Spontaneous malformations”
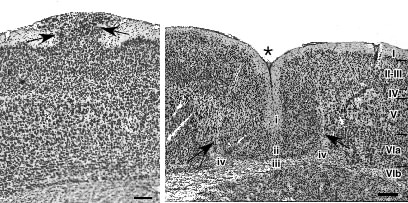 |
Figure 2 - Spontaneously occurring and induced minor neocortical malformations of the mice and rats. A. Photomicrograph of a molecular layer ectopia in an immune-disordered mouse (arrows). Bar = 400 µm. B. Photomicrograph of rat with an induced microgyria (asterisks). In comparison with the adjacent normal six-layered cortex (right), the microgyria is composed of four layers. Layer i is contiguous with the adjacent molecular layer. Layer ii is comprised of neurons normally found in layers II–III of normal cortex. Layer iii is a region of gliotic scarring and is the remnants of the layers IV–VIa of the normal cortex which are destroyed by the freezing injury. Layer iv, which is often discontinuous in the microgyria, is comprised of subplate cells. Bar = 200 µm. |
Summary
In this section, two different but related models of the anatomic malformations seen in the dyslexic brain have been presented. From this work, we have been able to glean a wealth of information that would not necessarily be amenable to study in the humans. We have evidence, for example, that these malformations are the result of injury occurring early in gestation and that they have a genetic component. This latter fact, combined with evidence from the human dyslexic suggesting a link with chromosomes 1, 2, 6 and 15 may yield more insights into the etiology of this disorder (88–93).
The parallels between the dyslexic and these animal models extend to behavior as well. Thus, while it is impossible to test animals for the phonological defects exhibited by dyslexics, these animals have measurable deficits in working memory — a difficulty also encountered in developmental dyslexics. In the following section, we present evidence suggesting that animals with neocortical malformations also have deficits in rapid auditory processing.
RAPID AUDITORY PROCESSING DEFECTS ASSOCIATED WITH SPONTANEOUS AND INDUCED MALFORMATIONS OF THE CORTEX
Spontaneous Malformations
As described above, minor developmental cortical malformations, including microgyria, are seen in the brains of dyslexics and in our animal models. Concomitant studies have shown that language-impaired (LI) children, a large subset of whom are later diagnosed as dyslexic, exhibit severe deficits in the discrimination of rapidly presented auditory stimuli, including phonological and non-verbal stimuli (i.e., sequential tones), specifically when total stimulus durations falls below 350 ms (34). Our animal models therefore provide a direct way to test the hypothesis that neocortical malformations are associated with alterations in neurophysiological responses to auditory stimuli.
In our first experiment (94), we tested 33 BXSB mice (12 ectopic and 21 non-ectopic) using a modified reflex modification paradigm. This consisted of the presentation of a pre-stimulus briefly preceding a startle-eliciting stimulus (SES)—a 105 dB white noise burst that causes mice to exhibit an acoustic startle reflex. When the pre-stimulus is detected, the amplitude of the whole-body, acoustic startle reflex is inhibited (“pre-pulse inhibition”). The extent of pre-pulse inhibition is related to the overall detectability of the pre-stimulus. Comparison of reflex amplitudes when a pre-stimulus is present (i.e., a cued trial) versus not present (i.e., an uncued trial) provides an objective measure of sensory detection (95). For the purpose of this study, we modified the task to allow us to assess threshold gap detection. Specifically, we presented a variable duration silent gap (0–100 msec) 50 msec before the SES, with the gap duration on each trial randomly selected. Trials occurred every 16 to 24 sec so as not to be predictable, and consisted of 75 dB continuous background white noise, the presentation of a silent gap, 50 msec of additional background white noise, followed by presentation of the SES (Figure 3 inset). A complete testing session contained trials with 0 (no gap), and 9 gap intervals ranging from 2–100 msec. For the purpose of statistical comparison, the 0 msec or “no gap” represented the “uncued” (baseline startle response) condition, while the “cued” conditions included gap durations of 2–100 msec.
In both non-ectopic and ectopic groups, a significant main effect of gap duration was observed—significant differences between uncued and cued response were evident at gap durations down to 5 msec, but not at 2 msec. Responses were then converted to percentages, specifically representing the cued response as a percentage of baseline (uncued) response for each subject, for each condition. If no advantage was conferred by a given gap duration (i.e., no detection), the cued response should approximate the uncued one (i.e., 100%). Interestingly, here we found a significant interaction between the presence or absence of ectopias and gap durations—ectopic mice were significantly impaired relative to non-ectopic mice at only the 5 msec gap condition. Ectopic and non-ectopic gap detection performance did not differ significantly at the remaining longer gap durations of 10 –100 msec (Figure 3).
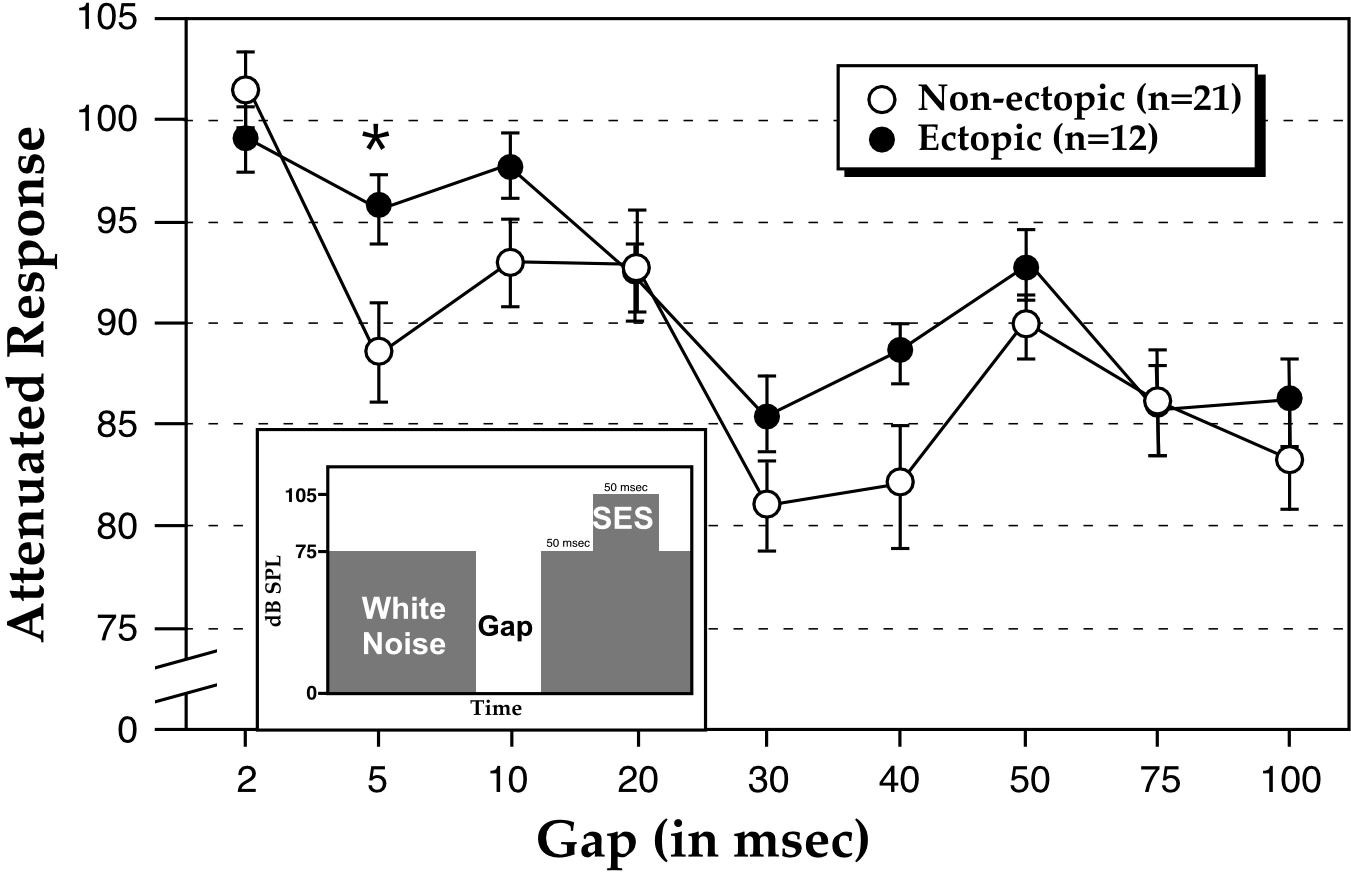 |
Figure 3 - Gap detection in ectopic and non-ectopic mice. Inset. Schematic representing gap detection protocol. The duration of the gap varied equally and randomly between the values of 0 (no gap), 2, 5, 10, 20, 30, 40, 50, 75 and 100 msec across 300 trials. SES = Startle Eliciting Stimulus. Main Scatterplot. Results from gap detection experiment. Ectopic BXSB mice are significantly impaired relative to non-ectopic BXSB mice at only the 5 msec gap duration. Attenuated Response is computed by dividing the startle response to cued trials (i.e., gap durations of 2, 5, 10, 20, 30, 40, 50, 75 and 100 msec) by the startle response to uncued trials (i.e., no gap condition) X100 (from 94) |
Thus, ectopic and non-ectopic male BXSB mice demonstrated significant gap detection in broadband white noise down to gap durations as low as 5 msec. These results approximate thresholds observed in other species including humans (96) and rats (97). At the shortest detectable gap (5 msec), however, ectopic mice showed, on average, significantly worse detection than non-ectopics. These results suggest that focal cortical malformations, in this case neocortical ectopias, are related to impairments in rapid auditory processing. The current results are consistent with data obtained from children with developmental dyslexia (98) wherein longer gap duration thresholds have been seen for affected individuals as compared to controls.
The experiment described above showed psychophysical differences between ectopic and non-ectopic mice. In our next experiment (99), we asked whether there were any neurophysiologic consequences of neocortical ectopias. We therefore implanted gold plated surface electrodes to record from primary auditory cortex in 10 BXSB mice (5 with and 5 without ectopias). Auditory stimuli were delivered to these unanesthetized animals in order to produce an auditory event related potential (AERP). The three stimulus protocols used in this study were designed to test changes in response to differences in stimulus timing and transition. In all protocols the initial stimuli were 10.5 kHz tones, 360 msec duration. This frequency was chosen because it is approximately at the peak of auditory sensitivity for mice. The initial 360 msec tone was followed by intervals of 12, 36, 72, 144, or 288 msec, during which there was either silence (10-gap-10), a 0.99 kHz tone (10–1–10), or a 5.6 kHz tone (10–5–10). Following this interval, a 120 msec second 10.5 kHz stimulus was delivered. Each of the 15 trial types (3 protocols, 5 intervals) was repeated 100 times with an intertrial interval of 600–700 msec (Figure 4A). It is important to note that data analysis of the AERP was performed without any knowledge of the histology status of BXSB mice.
The design of the study, therefore, was aimed at determining the conditions under which the second of the two stimuli would be detected (as measured by the AERPs). We found that the amplitude of AERPs to the second of the pair of 10.5kHz stimuli, measured from peak positivity to peak negativity, increased in amplitude as a function of increasing interval between the stimuli. Responses to second stimuli were minimal at 12 msec, but by 288 msec, responses to second stimuli consistently approached 100% of the amplitude of the first, regardless of whether or not animals had ectopias. As shown in Figure 4B and 4C, the entire response profile as a function of interval was not different between animals with or without ectopias for either the 10-gap-10 or 10–1–10 protocols.
In contrast, the amplitude of the second AERP in the 10–5–10 stimulus protocol differed significantly between mice with and without ectopias. Mice with ectopias showed small or no responses to second 10.5 kHz stimuli in the pair up to intervals of 72 msec, whereas at 36 msec intervals animals without ectopias showed responses to second 10.5 kHz stimuli that were approximately 50% the amplitude of the response to first stimuli. In fact, the responses to second 10.5 kHz stimuli was diagnostic for the presence or absence of ectopias—a non-expert was able to accurately categorize 9 of 10 animals as having ectopias or not based solely on the amplitude of the response to the second stimulus at short intervals. The effect of the ectopia on the amplitude of the response to the second stimulus is limited to the shortest intervals, and responses are identical at longer intervals. Therefore, the attenuation in cortical response is likely dependent upon an interaction of stimulus timing and contrast at stimuli transitions.
Taken together, these results suggest that neocortical ectopias in mice alter auditory processing in a fashion similar to that hypothesized to occur in dyslexics. Two stimulus features that are important variables for revealing neurophysiological alterations in language-impaired humans, timing delays and rapid changes in frequency (37), are also important variables for revealing ectopia-associated attenuation of AERPs in mice. Indeed, it appears that in animal models the presence of ectopias is associated with deficits in sensory processing when processing demands are highest. For example in the latter study, the deficit is apparent only at short intervals when a 5.6 kHz tone precedes the second 10.5 kHz stimulus and there is no deficit when preceded by either silence or 0.99 kHz tones. Similarly, in our first study discussed above, ectopic animals show an attenuation in startle reduction only at the shortest detectable gap durations. In sum, brains with focal neocortical malformations show impairment when rapid sensory processing demands are particularly high.
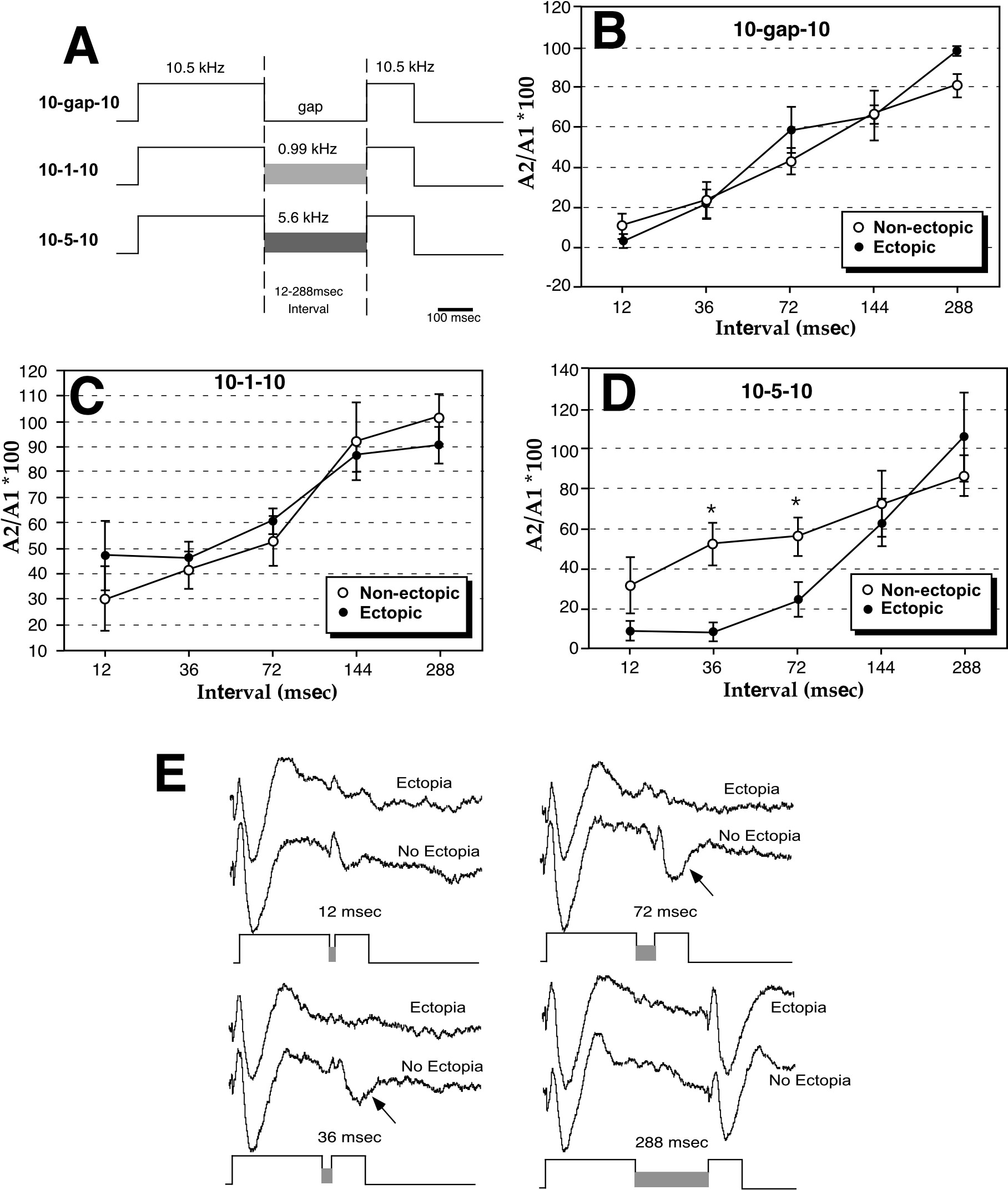 |
Figure 4 Auditory Event Related Potentials (AERPs) in ectopic and non-ectopic mice. A. The three stimulus protocols used in a study of AERP in ectopic and non-ectopic mice. In the first, two 10.5 kHz stimuli were separated by silent gaps (10-gap-10), in another by 0.99 KHz tones (10–1–10) and in a third by 5.6 KHz tones (10–5–10). The two stimuli were separated by 12, 36, 72, 144, or 288 msec intervals. All tones were 84 dB. B. Responses in the 10-gap-10 and 10–1–10 (C) protocols were nearly identical for all animals with and without ectopias. In each graph the % of the amplitude of the second to the first AERP (A2/A1 *100) is plotted against the interval between 10.5 kHz stimuli. D. The ratio of the amplitude of the second to the first AERP was significantly reduced in mice with ectopias for intervals of 36 and 72 msec. E. Grand average AERPs for mice with and without ectopias. Arrows indicate the near absence of AERPs to the second stimulus after 36 and 72 msec. |
Induced Malformations
Evidence from mice with spontaneous malformations of the neocortex strongly suggests a link between the presence of cortical malformations and defects in rapid sensory processing. It seems unlikely that the immune status of these animals plays a role in these results as both ectopics and non-ectopics are equally affected by the latter. Yet, the possibility of an interaction between the cortical malformations and the immune status of the subjects cannot be ruled out. To obviate this potential, if unlikely, confound, and to generalize these findings, we have tested rats with induced neocortical malformations on tasks involving auditory processing using paradigms both similar and distinct from those used in testing the mice with spontaneous neocortical malformations.
As described above, language-impaired (LI) children, a large subset of whom are eventually diagnosed with dyslexia, exhibit severe deficits in the discrimination of rapidly presented auditory stimuli, including phonological and non-verbal stimuli (i.e., sequential tones), specifically when total stimulus durations falls below 350 ms (34). This two tone sequence discrimination task which elicited these significant differences between non-reading-impaired children and those with either LI and/or dyslexia (34,100,101) was adapted to an operant conditioning go-no go target identification paradigm for rats (102). We sought to develop an animal model for impaired auditory temporal processing by exploiting the functional parallels between rats and humans in two-tone discrimination, and our ability to model some of the neuroanatomic anomalies seen in dyslexia. Toward that end, adult male rats with neonatally induced microgyria were tested in an operant paradigm for auditory discrimination of stimuli consisting of 2 sequential tones (103). Subjects were shaped to perform a go-no go target identification, using water reinforcement. Stimuli were reduced in duration from 540 to 249 ms across 24 days of testing. Discrimination indices were calculated from the difference, in msec, between mean latencies to respond to the target (hits) versus non-targets (false-alarms) for each subject across days. Results showed that all subjects were able to discriminate at longer stimulus durations. However, lesioned subjects showed specific impairment at stimulus durations of 332 ms or less, and were significantly depressed in comparison to shams. These results suggest a possible link between the neuropathologic anomalies observed in some dyslexics, and the rapid auditory processing deficits reported for LI subjects and dyslexics (34,100,101). More recent studies (104) have shown that female rats, unlike males, successfully perform the two-tone auditory discrimination task at all conditions, irrespective of whether or not they have induced microgyria (Figure 5). We have recently tested rats on a similar startle reduction paradigm to that described above for the mice. We found that rats with microgyria were unable to significantly discriminate two-tone stimuli presented with total stimulus durations less than 85 msec. These results lend strong support to the notion that rats with microgyria have difficulty processing rapidly presented auditory information (105).
We hypothesized that these differences in the rats’ ability to process rapidly presented auditory information might be reflected, as they may be in the dyslexic, by changes in the size of neurons in the primary auditory thalamic nucleus, the MGN (48). We therefore measured MGN neuron size, packing-density, and number in male and female rats with induced microgyria who were also tested for auditory discrimination learning. When we examined the MGN of our male subjects, there were more small and fewer large neurons in the microgyric as compared to controls animals. In contrast, there were no such differences in MGN neuron size distribution in the females (106).
 |
Figure 5 - Fast auditory processing is disturbed in male, but not female rats. Mean discrimination index for male (square) and female (circle) lesioned (filled) and sham (open) groups at the 4 stimulus duration conditions. Discrimination indices are mean scores over 6 days of testing at each condition. The numbers under each condition are total stimulus time (pre-tone/ISI/post-tone). |
Summary
Data derived from studies of subjects with both spontaneous and induced malformations appear to substantiate a relationship between the presence of malformations and problems with rapid auditory processing. Furthermore, there is evidence to suggest at least one anatomic substrate for these behavioral and physiological changes in animals with malformations. Our cumulative findings thus raise the question as to the mechanisms underlying this link between neocortical malformations and low-level disturbances in physiology, anatomy, and behavior. We have hypothesized that after injury causes these cortical malformations, reorganization of the cerebral cortex leads to propagation of effects throughout the brain in a top-down manner. Specifically, we posited that early injury (perhaps of a vascular origin) produces malformations with normal and abnormal efferent and afferent connectivity, which could provide the conduits for the propagation of cascading effects on structures thus connected to this region. In the following section, some recent investigations of the effects of cortical malformations on cerebrocortical connectivity are discussed.
Spontaneous malformations
As discussed above, immunohistochemical studies have shown an increased density of neurofilament-immunoreactive fibers radially oriented underlying ectopias (107). A follow-up DiI (a lipophilic tracer) study was designed to show definitively that these bundles contained afferent and efferent fibers from neurons in the ectopias. Small crystals of DiI were placed in the middle of an ectopia and the projections from the ectopia traced (108). In all cases there was a distinctive bundle of labeled fibers extending from the ectopic cells through the deeper layers of the cortex. This bundle of fibers then either entered the corpus callosum, the internal capsule or both (Figure 6A-D). Depending on the location of the ectopia within the somatosensory cortex, labeling was seen in appropriate thalamic nuclei. Cortico-cortical connections were also seen between ectopias in barrel cortex and both secondary somatosensory and primary motor cortices. Rarely was there any visible connections in the contralateral cortex.
The DiI labeling from non-ectopic cortex showed a distinctly different pattern of connectivity. In no case was there labeling of fiber bundles under the injection site in the controls. In comparison to the ectopias the non-ectopic cortex seemed to have less intense staining of the thalamic nuclei. The non-ectopic placements showed more fibers crossing the corpus callosum with distinctive patterns of connections in the contralateral cortex. These findings confirm the notion that layer I ectopias are anomalously connected by comparison to neurons in homologous cortex, which may underlie widespread dysfunction of brains containing ectopias.
Changes in connectivity with microgyria
Disturbed interhemispheric connectivity has been associated with a spontaneously occurring microgyrus in the rat cause spontaneously occurring microgyria are rare in experimental animal species, we injected in vivo tracers into the microgyria of adult animals. Specifically, we injected biotinylated dextran amine (BDA) into microgyric and matched control cases. In comparison to controls, animals with microgyria showed a decrease in efferent projections from the microgyric cortex to the opposite hemisphere (Figure 6E). Microgyria in the primary sensory cortex (Par1) had abnormal efferent connections to secondary somatosensory cortex (Par2) of the opposite hemisphere (Figure 6F). Injection of BDA into the homologous area of the undamaged hemisphere highlighted aberrant projections into frontal and Par2 cortex of the affected hemisphere (109). There were almost no thalamocortical or corticothalamic projections between the ventrobasal complex and the microgyrus itself. Interestingly, a dense plexus of thalamocortical fibers was often noted at the border between the malformed and normal cortex, an area shown to be important in the generation of epileptogenic discharges (110,111).
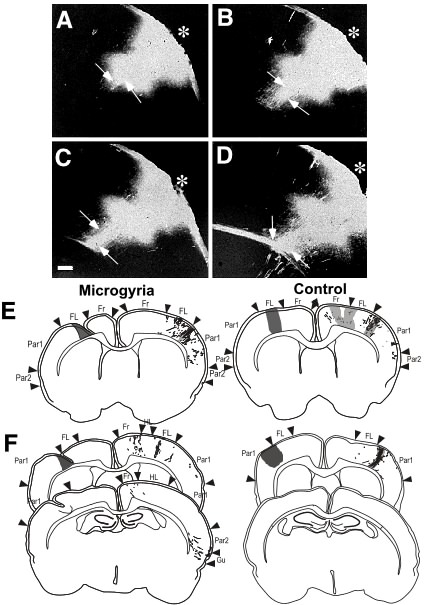 |
Figure 6 A-D. Connectional changes in ectopic mice. Images illustrating a bundle of fibers from an ectopia in FL cortex of an immune-disordered mouse (arrows). The bundle is seen entering the corpus callosum in panels C and D. Bar = 200 µm. E. Connectional changes in Rats. Tracing of darkfield photomontages of injection into a microgyrus (dark gray) located in the forelimb region of the right hemisphere. Retrogradely labeled cells are represented as filled circles, and anterogradely labeled fibers are traced. Dense patches of projections are represented by light gray-filled regions. Patterns of projections to the left hemisphere are notable for an absence of the normal homotopic projections to FL as seen in the matched control, In the microgyric case, the densest projections are located in Par1 and are far less dense than those in the control. F. Heterotopic projections of Microgyric Animals. Tracing of darkfield photomontages of injection into a microgyrus located in the border between FL and Par1 of the right hemisphere of (dark gray) illustrating retrograde and anterograde connections. When compared to the matched control there is a distinct decease in density of projections to the homotopic cortical region. In addition, heterotopic projections to Fr (bottom section) and Par2 and Gu (top section) can be seen in the microgyric case but not in the control. Abbreviations: Fr = Frontal cortex (including Fr1, Fr2, and Fr3); FL = Forelimb area of parietal cortex; HL = Hindlimb region of parietal cortex; Par1 = Primary parietal cortex; Par2 = Secondary parietal cortex; Gu = Gustatory cortex. |
Summary
We had previously shown that minor malformations of the cerebral cortex were associated with defects in low-level physiologic, anatomic, and behavioral systems. What remained unexplained was how to damage to high-level structures could affect these low-level substrates. We had hypothesized that after injury causes these malformations, reorganization of the cerebral cortex leads to propagation of effects throughout the brain in a top-down manner. One conduit for propagation of changes could be neuronal connections. Connections between the injured area and the thalamic target could be (1) direct, (2) developmentally transient, (3) polysynaptic, or (4) new connections produced by the injury. Our research does not speak directly to this issue, but we know from other sources that the maintenance of transient connections is one distinct possibility (112–114). Furthermore, widespread brain reorganization has been reported following a variety of developmental injuries. In hamsters, for example, neonatal lesions of the superior colliculus result in altered retinal projections (115–117).
It is possible, therefore, that early injury to the cortical plate causes minor neocortical malformations with normal and abnormal efferent and afferent connectivity, which could provide the conduits for the propagation of cascading effects on structures thus connected to this region. The abnormal connectivity could theoretically be the result of the pathological maintenance of normally transient connections or the formation of novel connections. Although propagation of changes along standard connections would be easily traced, changes propagated via transient or anomalous connections require special experimental demonstration.
The work discussed in this chapter has pointed to a number of areas where research into the biological substrates of developmental dyslexia has been aided by the use of animal models. From examination of animals with either induced or spontaneous malformations, we have learned more about the etiology of these malformations as well as their effects on brain organization. In addition, behavioral testing has pointed to some intriguing parallels with the non-linguistic deficits seen in dyslexia and other language impairments. In the case of the spontaneous malformations, for example, there is evidence that ectopic animals have working memory deficits.
Perhaps most important for the topic at hand, however, is that the examination of these animal models has enabled us to link together the presence of minor malformations of the neocortex and defects in rapid auditory processing. We know that the induction of microgyria into an otherwise normal cortex profoundly disturbs the ability of that animal to process auditory information when it is presented in rapidly. Similarly, mice with spontaneous malformations of the neocortex also have difficulties processing rapidly presented auditory information, and this difference is manifest at the physiologic level. This supports the notion that this defect in rapid sensory processing may be caused by a top-down disruption of cerebral architecture induced by the early injury. The mechanisms for this disruption are hypothesized to be the result of connectional reorganization—a hypothesis that is supported by recent findings.
Although there are obvious and important limits to the types of questions that we can ask non-human animals with respect to developmental dyslexia, there are similarities in the basic biological substrates that may underlie this disorder that allow and encourage the use of animal models. It is hoped that as we learn more about the basic anatomical, behavioral, and cognitive aspects of this disorder that other similarities will become evident and allow researchers to converge on a more complete understanding of this disorder.
ACKNOWLEDGMENTS
This work was supported, in part, by PHS grant HD20806.
The authors wish to thank Victor H. Denenberg, Amy Herman, Christine Brown, Wendy Gray, and Paula Tallal for their contributions to the work reported here. Acknowledgments also go to Antis Zalkalns, Lisa Stone Garcia, and Heinz Windzio for technical assistance.