Mapping the Best mutation on mouse Chromosome 16: a model for human optic atrophy
D.S. Rice,1 R.W. Williams,1 P. Ward-Bailey,2 K.R. Johnson,2 B.S. Harris,2 M.T. Davisson,2 D. Goldowitz1
1The University of Tennessee, Center for Neuroscience and Department of Anatomy and Neurobiology, 875 Monroe Avenue, Memphis, Tennessee 38163, USA
2The Jackson Laboratory, Bar Harbor, Maine 04609, USA
Received: 20 March 1995 / Accepted: 5 April 1995
Autosomal dominant optic atrophy (OPA1) is the most common form of hereditary optic atrophy in humans, with an incidence of 1:50,000 (165500; GDB 1995). A recent study has localized OPA1 to Chromosome (Chr) 3 between q28-qter (Eiberg et al. 1994). OPA1 is characterized by a loss of visual acuity, deficits in color vision, and scotomas of varying size (Eliott et al. 1993). Retinas of patients with OPA1 have a reduction in the number of retinal ganglion cells and a decrease in myelin content in the optic nerves, chiasm, and tracts (Johnston et al. 1979; Kjer et al. 1983). Neurons that are in the main target of the retinal ganglion cell projection, the dorsal lateral geniculate nucleus, are also atrophic (Kjer et al. 1983). Other cell populations in the retina of humans with OPA1 appear to be normal (Johnston et al. 1979; Kjer et al. 1983). OPA1 is a dominant mutation, but the expression of the phenotype is highly variable both within and among families (Kline and Glaser 1979). The loss of visual acuity and the atrophy of the optic nerves often varies between right and left sides (Kline and Glaser 1979; Kjer et al. 1983).
Recently, we have identified a striking abnormality in optic nerves of mice that are heterozygous for the spontaneous mutation belly spot and tail (Bst). Bst is a semi-dominant, homozygous lethal mutation that arose in the inbred strain C57BLKS (BKS; previously denoted C57BL/Ks). Heterozygous mice have a kinky tail, white feet, and a white spot at the ventral midline. In approximately 50% of the Bst/+ mice, there is a reduction or a complete absence of the pupillary light reflex in one or both eyes (Rice et al. 1993). This neurological phenotype is associated with a unilateral or bilateral atrophy of the optic nerves. As in humans with OPA1, the severity of the atrophy of the optic nerves is highly variable--ranging from a slight reduction in the number of ganglion cell axons in one optic nerve to a complete elimination of both optic nerves. The surface area of the retina and the appearance of the inner and outer nuclear layers are qualitatively normal (Rice et al. 1993).
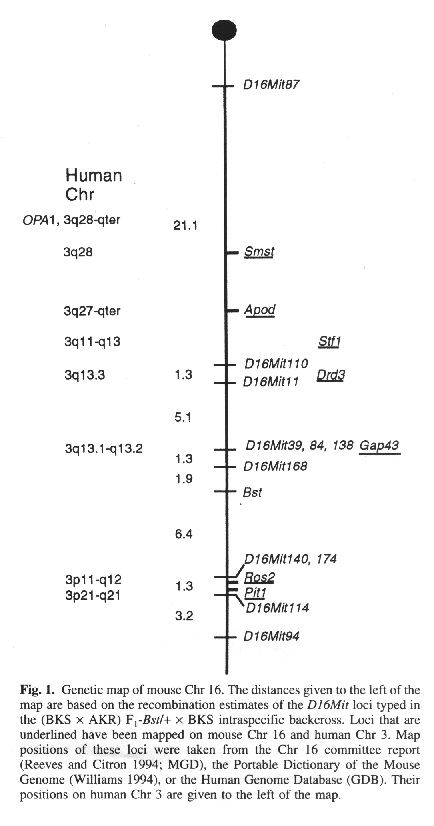
The Bst locus has been mapped previously as the distal-most locus of a three-point cross in relation to Igl1 (immunoglobin lambda-I) and md (mohaganoid) on Chr 16 (Epstein et al. 1986). Harris et al. (1989) subsequently mapped Bst in a two-point cross with Sod1 (superoxide dismutase- 1). Collectively, these studies place the Bst locus 25 to 42 cM distal to the centromere and proximal to Sod1. This region of mouse Chr 16 is conserved in human Chr 3 (Reeves and Citron 1994). Given the marked phenotypic similarity of retinal phenotypes between OPA1 and Bst and the chromosomal homology, we have generated a higher resolution map of Bst on Chr 16 using an intraspecific backcross. F1
hybrids were produced by crossing BKS-Bst/+ females to AKR males. The (BKS x AKR)F1-Bst/+ males and females were crossed to wildtype BKS, generating a total of 157 backcross progeny. These progeny were phenotyped for Bst by inspecting the tail for kinks and the belly for white hairs. Genomic DNA was isolated for the analysis of simple sequence length polymorphisms (SSLP). A total of 11 primer pairs recognizing SSLP loci were used to map Bst more precisely. PCR reactions were carried out as described by Dietrich and associates (1992) with two modifications: (i) the Taq DNA polymerase concentration was doubled (0.5U/rxn), and (ii) 30, instead of 25 cycles, were run. PCR products were separated on 7% nondenaturing polyacrylamide gels and stained with ethidium bromide. Recombination frequencies were analyzed with the program Map Manager v2.51 (Manly and Elliott 1991).

The results of the haplotype analysis establish the following order among loci with distances in cM ± standard error: D16Mit87-21.1 ± 6.6-D16Mit110-1.3 ± 1.3-D16Mit11-5.1 ± 1.8-D16Mit138, D16Mit84, D16Mit39-1.3 ± 0.9-D16Mit168-1.9 ± 1.1-Bst-6.4 ± 2.0-D16Mit14O, D16Mitl74-1.3 ± 0.9-D16Mit114-3.2 ± 1.4-D16Mit94. Thus, according to the placement of these markers on the consensus map for mouse Chr 16 (Reeves and Citron 1994), Bst lies approximately 39 cM from the centromere (Fig. I).
The percentage of mutants in the backcross (33.8%) is less than the 50% expected for a dominant Mendelian trait. There are two possible explanations. First, the penetrance of the mutation may be decreased in the backcross progeny (see Epstein et al. 1986). If the paucity of Bst/+ mutants in the backcross is reduced because of incomplete penetrance, then flanking markers should still exhibit normal 1:1 segregation. However, in our backcross, markers linked to the Bst locus exhibit a marked segregation distortion, consistent with our Bst genotype assignments (Fig. 2). Second, the in utero survival rate of the Bst heterozygote may be compromised. The mean litter size is reduced in the backeross compared with the intercross (6.0 vs. 7.8), and the percentage of Bst/+ mutants is reduced in backcross litters (Table I). Furthermore, Bst heterozygotes on the C57BLKS inbred background are not as viable (mean litter size = 4.6) as heterozygous mice whose genetic background is a mosatc of AKR and C57BLKS alleles. Thus, the disparity in the BKS alleles in the backcross is probably the result of the in utero elimination of Bst/+ mutants. In a related study, we have found in utero death associated with a high incidence (about 30%) of exencephaly in crosses using Bst/+ mice (Rice et al. 1993).
We have mapped the Bst locus to a region of mouse Chr 16 that is conserved in human Chr 3. Some of the loci on human Chr 3 that are also located on mouse Chr 16 include: apolipoprotein d (APOD), pituitary transcription factor (PIT1), dopamine D3 receptor (DRD3), preprosomatostatin (SMST), stefin 1 (STFI), R0S2, and growth-associated protein (GAP43; Fig. I). Gap43 has recently been shown to be important for the growth of retinal ganglion cell axons at the optic chiasm in the mouse (Strittmatter et al. 1995). However, on the basis of the current map positions, GAP43 (which maps to human Chr 3q13.1-q13.2; Naylor et al. 1994) does not appear to be a candidate for OPA1 although it cannot be ruled out as a candidate for the Bst mutation.
OPA1 and Bst have many similarities. Both mutations are inherited as dominant phenotypes with variable expressivity, and both appear to target retinal ganglion cells. It has been postulated that the OPA1 mutation results in a primary degeneration of the cells in the ganglion cell layer and, subsequently, in atrophy of the optic nerves (Kjer et al. 1983). The decrease in retinal ganglion cells in the Bst/+ mouse is evident as early as the day of birth (Rice et al. 1993) and before the onset of naturally occurring ganglion cell death (Williams et al. 1990).
The mechanisms responsible for the congenital decrease to ganglion cell number in humans with the OPA1 mutation and in Bst/+ mutant mice are unknown. Comparison of the order of homologous loci in the mouse and human chromosomal maps for this region suggests that OPA1 and Bst map to different regions of the conserved segment. Therefore, they either may not be mutations in the same gene or the gene order may differ between the mouse and human chromosome within these conserved segments. Despite this caveat, the remarkable similarities in the atrophy of the ganglion cell population between OPA1 and Bst make the Bst mutant mouse a good model to study the abnormal development of the ganglion cell population.
Acknowledgments. We thank Janice Hyatt for assistance with genomic DNA isolation. This work was supported by National Institutes of Health (NIH) Grant NS EY-09586 (D. Goldowitz), NIH Neuroscience training grant NS-07323 (D.S. Rice), NIH Grant PO1 RR01183, and Cancer Core Grant CA34196 (B.S. Harris, P. Ward-Bailey, KR. Johnson, MT. Davisson).
References
Dietrich, W.F., Katz, H., Lincoln, SE., Shin, H.-S., Friedman. J., Dracopila, N., Lander, E.S. (1992). A genetic map of the mouse suitable for typing intraspecific crosses. Genetics 131, 423-447.
Eiberg, H., Kjer, B., Kjer. P., Rosenberg, T. (1994). Dominant optic atrophy (OPA1) mapped to chromosome 3q region. I. Linkage analysis. Hum. Mol. Genet. 3, 977-980.
Eliott, D., Traboulsi, E.I., Maumenee. I.H. (1993). Visual prognosis in autosomal dominant optic atrophy (Kjer type). Am. J. Ophthalmol. 115, 360-367.
Epstein, R., Davisson, M.T., Lehmann, K., Akeson. E.C., Cohn, M. (1986). Position of Igl-1, md, and Bst loci on chromosome 16 of the mouse. Immunogenetics 23, 78-83.
GDB, Human Genome Database. (1995). The Johns Hopkins University Bininformatics Web Server (URL:http//gdbwww.gdb.org).
Green, M.C., Witham, B.A. (1991) Handbook on Genetically Standardized JAX Mice, 4th ed. (Bar Harbor, ME: The Jackson Laboratory).
Harris, B.S., Cook, S., Davisson, M.T. (1989). Mouse News Lett. 84, 90.
Johnston, P.B., Gaster, R.N., Smith, V.C., Tripathi, R.C. (1979). A clinicopathologic study of autosomal dominant optic atrophy. Am. J. Oph thalmol. 88, 868-875.
Kjer, P., Jensen, O.A., Klinken, L. (1983). Histopathology of eye, optic nerve and brain in a case of dominant optic atrophy. Acta Ophthalmol. 61, 300-312.
Kline, L.B., Glaser, J.S. (1979). Dominant optic atrophy. The clinical profile. Arch. Ophthalmol. 97, 1680.
Manly, K.F., Elliott, R.W. (1991). RI manager, a microcomputer program for analysis of data from recombinant inbred strains. Mamm. Genome 1, 123-127.
Mouse Genome Database (MGD). (1995). Mouse Genuine Informatics Project, The Jackson Laboratory, Bar Harbor, Me. World Wide Web (URL:http//www.informatics.jax.org).
Naylor, S.L., Buys, C.H.C.M., Carritt, B. (1994). Report on the fourth annual workshop on human chromosome 3 mapping. Cytogenet. Cell Genet. 65, 2-34.
Reeves, R.H., Citron, M.P. (1994). Mouse Chromosome 16. Mamm. Genome 5 (suppl.), S229-S337.
Rice, D.S., Williams, R.W., Davisson, M.T., Harris, B., Goldowitz, D. (1993). A new mutant phenotype of retinal ganglion cell disgenesis discovered in the mouse. Society of Neuroscience Abstr. 19, 51.
Strittmatter, S.M., Fankhauser, C., Huang, P.L., Mashimo, H., Fishman, M.C. (1995). Neuronal pathfinding is abnormal in mice lacking the neuronal growth cone protein Gap-43. Cell 80, 445-452.
Williams, M.A., Pinon, L.G.P., Linden, R., Pinto, L.H. (1990). The pearl mutation accelerates the schedule of natural cell death in the early postnatal retina. Exp. Brain Res. 82, 393-400.
Williams, R.W. (1994). The portable dictionary of the mouse genome: a personal database for gene mapping and molecular biology. Mamm. Genome 5, 372-375. World Wide Web (URL:http//www.nervenet.org).
Mammalian Genome 6: 546-548 (1995).
Correspondence to: D. Goldowitz
Since 16 June 99