![]()
Note to the reader: This is a revised edition of a paper published in Developmental Brain Research (1992;67:285–91). The definitive original print version is available from Elsevier Press.
New figures, text, and links have been incorporated into the revision. Revised HTML (http://www.nervenet.org/netpapers/Rosen/Puncture92/Puncture.html) copyright ©1999 by Glenn D. Rosen
INDUCTION OF MOLECULAR LAYER ECTOPIAS BY PUNCTURE WOUNDS IN NEWBORN RATS AND MICE
Glenn D. Rosen, Gordon F. Sherman, Judy M. Richman, Lisa V. Stone, & Albert M. Galaburda
Dyslexia Research Laboratory, Beth Israel Deaconess Medical Center; Division of Behavioral Neurology , Beth Israel Deaconess Medical Center, 330 Brookline Avenue, Boston MA 02215; Harvard Medical School, Boston, MA 02115.
All correspondence should be addressed to:
Glenn Rosen, Ph.D.
Department of Neurology
Beth Israel Deaconess Medical Center
330 Brookline Ave
Boston, MA 02215
Email:grosen@caregroup.harvard.edu
Molecular layer ectopias spontaneously occur in immune-disordered mice, and the accompanying paper demonstrates that these ectopias are associated with a break in the external glial limiting membrane and with distortion of radial glial fibers at birth. It was hypothesized that injury to the developing neocortex is the main etiologic event for molecular layer ectopias. To test this hypothesis, puncture wounds were made on the surface of the cerebral cortex of newborn rats and mice. These wounds produced, in adulthood, molecular layer ectopias similar in appearance to those seen spontaneously in immune-disordered mice. Further, these ectopias show similar distortions of radial glial fibers during development, and of neurofilaments in adulthood. This work supports the notion that injury could be a factor in the production of molecular layer ectopias.
Cerebrocortical microdysgenesis, including molecular layer nests of neurons is present in the brains of dyslexic individuals examined at autopsy(1–4). Because of the suggested connection of immune disorders and learning disabilities (5–7), we examined the brains of immune-disordered mice for evidence of pathology (8–10) and found cerebrocortical microdysgenesis, characterized by ectopic collections of neurons with underlying dysplasia in the molecular layer of the neocortex, in up to 50 percent of the cases examined. There are disturbances of the local neuropil and connectivity associated with these ectopias as revealed by an increase in certain subpopulations of neurons (11), and a striking distortion of neuronal fibers and dendrites within and below the ectopias as revealed by neurofilament staining (12).
More severe forms of cerebrocortical microdysgenesis, namely microgyria, can be induced in rats by placing a freezing probe on the skull of newborns (12–15). This induced microgyria resembles human four-layered microgyria and generally consists of a fused molecular layer which comprises a sulcus (layer i of the microgyria), a cell dense layer contiguous with layers II-III of intact neocortex (but less differentiated; layer ii), a lamina dissecans (layer iii), and a layer of cells contiguous with layer VIb of adjacent, intact neocortex (layer iv). Molecular layer ectopias are sometimes seen, most often in the periphery of the neuropathologic lesion, and these ectopias show distorted neurofilament processes similar to those seen in spontaneous ectopias in the mouse (14,16). In the accompanying paper, we report that spontaneous molecular layer ectopias are associated with rupture of the external glial limiting membrane and an altered radial glial fiber network (17). It is hypothesized that this rupture is the result of injury to the glial limiting membrane before or during neuronal migration. In this study we test this hypothesis by mechanically disrupting the external glial limiting membrane of mice and rats in the hope of inducing cerebrocortical microdysgenesis.
Protocol
Two litters each of Wistar rats (Charles River, Wilmington, MA) and DBA/2J mice (Jackson Labs, Bar Harbor, ME) were used in the experiment. On the day of birth (P0), selected pups from the litters were subjected to puncture wounds of the neocortex (see below). Rats were sacrificed at either P0, P1, P2, or in adulthood (> P60) by transcardial perfusion with 0.9% saline followed by 4% paraformaldehyde in 0.1 M phosphate buffer under deep anesthesia (hypothermia or Pentobarbital depending on the age of the animal). DBA mice were sacrificed under deep anesthesia (hypothermia or ether) either 2 hours after the puncture wound, or at P1 or P41. The early lesioned mice were perfused only with 0.9% saline while the P41 mice were perfused identically to the adult rats. Following perfusion, the brains were removed from the skulls (rats and adult mice) or left intact (P0 and P1 mice) and postfixed for 24 hours, except for the P0 and P1 mice whose crania were postfixed for 6–8 hours. The brains were then placed in a 10% sucrose/0.1 M phosphate buffer overnight after which they were placed into 30% sucrose in 0.1 M phosphate buffer. When the brains had sunk, they were serially sectioned coronally at 30 µm on a freezing microtome. A series of every 5th section was mounted onto glass slides, stained with either Thionin or cresyl violet for Nissl substance and adjacent sections immunohistochemically stained for radial glial fibers (vimentin), astrocytes (GFAP), or neurofilament. Sections were examined under light microscopy.
Puncture wounds
A small incision was made in the skin over the midline of the cerebral hemisphere of a pup under hypothermia-induced anesthesia. The skull overlying either the left or right hemisphere was exposed, and a 25 gauge hypodermic needle was manually inserted through the skull into the underlying cerebral cortex. In the rats that were to be sacrificed in adulthood, 5–6 puncture wounds were performed, whereas only 2 puncture wounds per hemisphere were attempted in the mice and the rats sacrificed within 96 hours of the injury. These puncture wounds were placed at a distance sufficient to avoid confusion on subsequent histology. Following the puncture wounds, the skin was sutured shut, the pups warmed under a heat lamp, and the pups returned to their mothers.
Histology
Nissl
Sections were stained either with 0.05% Thionin or 0.5% cresyl violet following standard procedures.
Vimentin
We used vimentin to stain for radial glial fibers in animals sacrificed within 96 hours of the puncture wound. Free-floating sections were rinsed twice in phosphate-buffered saline (PBS; pH 7.4) for five min each and transferred to a buffered 0.6% hydrogen peroxide solution in order to block staining of endogenous peroxidases. The sections were rinsed twice in PBS and incubated overnight at 4°C in a 1/500 dilution of mouse anti-vimentin immunoglobulin (monoclonal antibody from Boehringer Mannheim, Indianapolis, IN). The vehicle (diluent) for all antibody incubations was 3% rabbit serum in PBS and 0.3% triton X-100.
Sections were then placed into a solution containing the linking antibody (rabbit anti-mouse immunoglobulin—Dakopatts Z259—diluted 1/20) at room temperature for two hours. The sections were rinsed twice with PBS and exposed to a 1/250 dilution of mouse peroxidase anti-peroxidase (Dakopatts B650) at room temperature for two hours. The tissue was rinsed twice in PBS and then twice in 50 mM Tris buffer (pH 7.6) and developed using 0.05% diaminobenzidine and 0.01% hydrogen peroxide diluted in Tris. After rinsing with Tris, sections were mounted on chrome-alum coated slides, dehydrated, counterstained with Methyl Green/Alcian Blue, coverslipped with Permount.
Neurofilament
The procedure for neurofilament staining is identical to that for vimentin with the exception of the dilution of the antibody (monoclonal antibody to the 68 kDa subunit of neurofilament from Boehringer Mannheim, Indianapolis IN), which is 1/33.
Glial Fibrillary Acidic Protein (GFAP)
GFAP was used to visualize the distribution of astrocytes around the puncture wound. Free-floating sections were rinsed twice in phosphate-buffered saline (PBS; pH 7.4) for five min each and transferred to a buffered 0.6% hydrogen peroxide solution in order to block staining of endogenous peroxidases. Sections were placed into a 1/25 solution of primary antibody (Incstar, Stillwater, MN) overnight at 4°C. The vehicle (diluent) for all antibody incubations was 3% goat serum in PBS and 0.3% triton X-100. The next day, sections were transferred into a biotinylated goat anti-rabbit immunoglobulin solution (Vector Laboratories, Burlingame, CA) diluted 1/60 for two hr at room temperature. After two washes in PBS, the sections were placed into ABC (Vector Laboratories) for two hr at room temperature. The tissue was rinsed twice in PBS and twice in 50 mM Tris buffer (pH 7.6) and developed, dehydrated, counterstained, and coverslipped.
Examination of the brains 24–48 hrs after the puncture wound revealed a variable degree of damage to the neocortex depending on the site of injury. In some cases, damage to layers I-III could be observed whereas in others the damage extended to layer V (Fig. 1, 2a). In a few cases the probe entered the white matter. In all cases, collections of ectopic neurons could be seen in the molecular layer (Fig. 1, 2a). Reactive astrogliosis could be seen within 48 hours after the puncture wound (Fig. 2c).
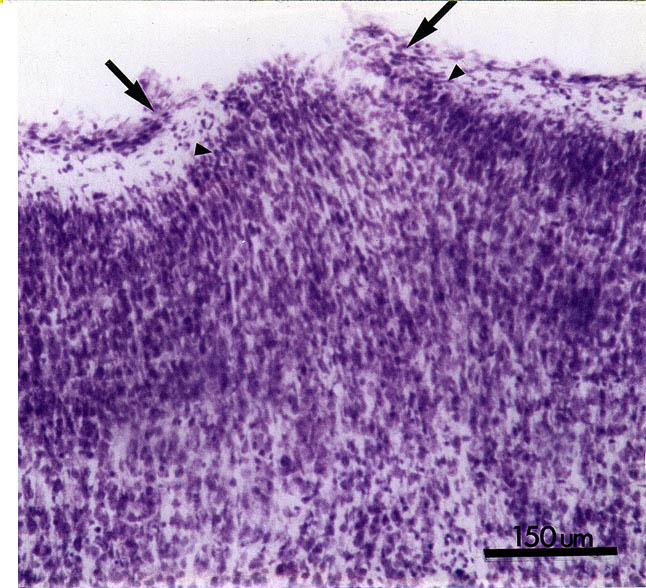 |
Figure 1. Nissl-stained section of the neocortex of a rat 24 hours after being subjected to puncture wound on the day of birth (P0). Note break in the pial membranes (arrows) and the appearance of neurons in the molecular layer. Bar = 150 µm.
|
 |
Figure 2. A. Nissl-stained section of the neocortex of a rat 48 hours after being subjected to a puncture wound on P1. As with Fig. 1, note the break in the in the pial membranes (arrows) and collections of neurons in the molecular layer (arrowheads). Bar = 500 µm. B. Section adjacent to panel A immunohistochemically stained for radial glial fibers with vimentin (arrowheads for orientation with panel A). Bar = 250 µm. C. Section adjacent to panel B immunohistochemically stained with GFAP demonstrating reactive astrogliosis in the area of the damage (arrows). Bar = 500 µm. D. Higher powered photomicrograph of panel B showing disrupted radial glial fibers. Some of these have swelled protuberances on their distal portions which have some characteristics of radial glial growth cones (arrows). Bar = 100 µm
|
Vimentin staining revealed a break of the external glial limiting membrane and damage to radial glial fibers directly beneath the injury within 48 hours (Fig. 2b,d). Specifically, there was an absence of intact fibers directly beneath the wound although there were swollen protuberances in the distal portion of these fibers which have some characteristics of radial glial growth cones(18; Fig. 2d). Those radial glial fibers that were intact curved toward the periphery of the damaged area and were attached to the intact portions of the glial limiting membrane in a manner similar to that seen in spontaneous ectopias (Fig. 2d). There were no changes in the radial glial fiber network outside the area of focal damage. It should be noted that there is significant restructuring of the astro-glial lineage during the postnatal period (19). Our material, however, did not lend itself to examination of any subtle changes that may have occurred in the normal course of this restructuring.
When examined in adulthood, puncture wounds of the neocortex were associated with molecular layer ectopias (Fig. 3a,b). These consisted of nests of neurons in layer I which, in general, were cylindrical in shape and did not differ in their mediolateral extent from layer II to the pial surface. Areas of neuronal dysplasia were sometimes, but not uniformly, noted in the area of the puncture wound (Fig 3b). These dysplastic areas normally were seen in layers II-IV. Neurofilament staining revealed disturbances of neuronal processes associated with cell-free areas underlying the area, which were similar to those seen in association with spontaneous neuronal ectopias in immune-disordered mice (Fig. 4a,b,c). Occasionally, disruption of the infragranular layers beneath the ectopias was noted (Fig 4a), but this was not a consistent finding in all cases.
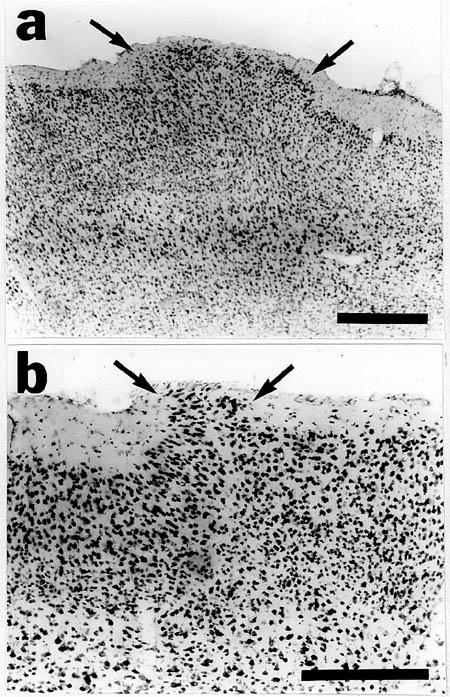 |
Figure 3. Nissl-stained neocortex of adult rat (A) and mouse (B) following a puncture wound at P0 demonstrating molecular layer ectopias (arrows). Bars = 500 µm (A) and 250 µm (B).
|
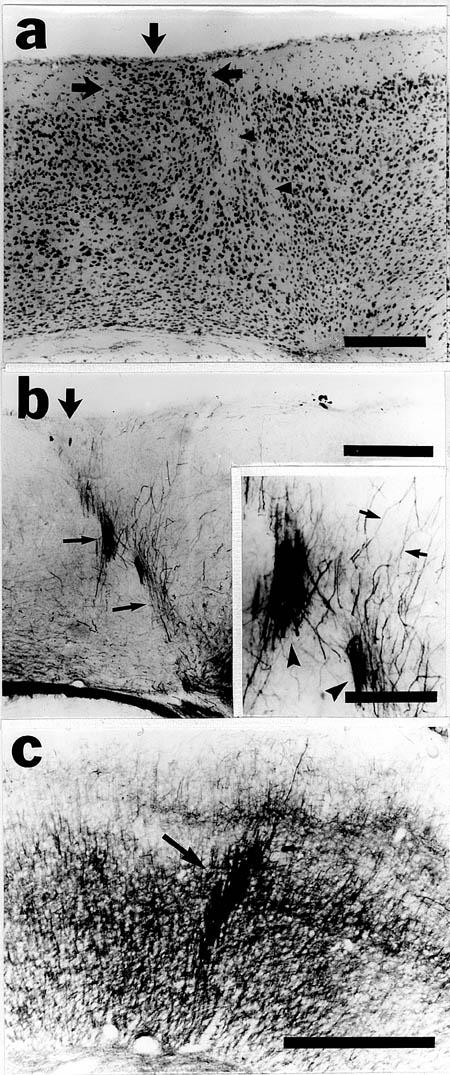 |
Figure 4. A. Nissl-stained neocortex of adult mouse following a puncture wound at P0 demonstrating molecular layer ectopia (arrows) and dysplasia of the underlying neocortex (arrowheads). B. Section adjacent to panel A immunohistochemically-stained for 68 kD neurofilament demonstrating aberrant bundles of neurofilament-positive fibers (thin arrows) associated with the ectopia (large arrow). These are similar in appearance to those seen in mice with spontaneous ectopias (panel C) . Inset shows these bundles in greater detail (arrowheads) as well as individual neurofilament-positive fibers (arrows). Bars for panels A, B, and C = 250 µm, Bar for inset to panel B = 100 µm
|
Induction of molecular layer ectopias
Molecular layer ectopias can be induced in rodent brains by puncture wounds during the newborn period. These ectopic collections of neurons are remarkable for their resemblance to spontaneously occurring ectopias in immune-disordered mice and in humans. Thus, aberrant patterns of neurofilament staining are seen in both the spontaneous and induced forms of neuropathology. In addition, vimentin immunostaining in mice, in association with puncture wounds, revealed breaks in the external glial limiting membrane which were similar to those seen associated with ectopias spontaneously in NZB mice. These results support the hypothesis that the formation of molecular layer ectopias may be the result of damage to the external glial limiting membrane occurring sometime during the course of neuronal migration (14, 15, ,17, 20). Alternatively, since the entire superficial plexiform layer is disrupted by the puncture wound, it could be that disruption of other components in this layer that normally act to provide a barrier against migrating cells are implicated in the formation of these ectopias (21–23).
Although similarities exist between spontaneous and induced molecular layer ectopias, there are some differences. In the NZB mouse, the nests of ectopic neurons most often take on a "mushroom-like" appearance with a narrow base and wider spread of neurons near the pial surface (8–10). In the induced case, however, the collections of ectopic layer I neurons appear cylindrical with no difference in the mediolateral extent from the base to the pial surface. We believe this difference to be minor, however, as the induced molecular layer ectopias resemble many of those seen in humans and mice (1–4, 24).
The importance of timing
The current experiment does not explore in depth the issue of timing of the insult in producing molecular layer ectopias, specifically the age at which it can occur and the length of time that the brain needs to be exposed to the injurious agent. In the case of spontaneous cerebrocortical microdysgenesis, the break in the external glial limiting membrane can be seen at birth. Yet it is not known whether the insult can occur earlier in gestation, how long the external membrane remains ruptured, or whether a disturbance of this membrane occurring after the end of neuronal migration would also cause the formation of molecular layer ectopias. Experimental work in induced microgyria (12–15) showed that a true four-layer anomaly cannot be produced after completion of neuronal migration, which would suggest that post-migrational production of molecular layer ectopias is also unlikely. This specific hypothesis needs to be tested directly.
In order to explore more fully the issue of timing, it would be important to perform puncture wounds at earlier and later ages than those undertaken in the current study. While there are no reports of prenatal puncture wounds, the effect of puncture or stab wounds in adulthood has been examined, although the interest in the effect of puncture wounds was unrelated to the creation of morphologic cerebrocortical anomalies (25–31). Thus, puncture or stab wounds result acutely in increased macrophage activity in the area of damage(26, 28) and chronically to the formation of a glial scar (28). Protein synthesis is inhibited in the brains of adult mice undergoing cerebral puncture wounds of a similar type to those used here, although there was no inhibition of protein synthesis in P7 or P8 mice (27, the youngest age to receive a puncture wound).
Etiology of molecular layer ectopias during neocortical migration
The pathogenesis of molecular layer ectopias in humans has long been the subject of speculation. Some have contended that these anomalies are the result of primary problems with neuronal migration, but most workers now believe it is secondary to damage around this time period (32–35) damage. The present experiment supports this position, but the question remains as to the mechanics of ectopia formation following damage during neocortical migration. In the case of normal neuronal migration to the neocortex, young neurons in the ventricular zone attach to radial glial cells to aid their journey to the cortical plate. Upon reaching the external glial limiting membrane and the superficial plexiform layer, these neurons cease their migration and detach from the radial glial cells. These neurons maintain their position while subsequently migrating neurons arrive in more superficial locations (18, 36–39). In the case of damage (like the type detailed in this paper), it is possible that those neurons associated with the damaged radial glial cells detach. Without an intact external glial limiting membrane (and/or damaged superficial plexiform layer) there is no barrier to keep these neurons from either unguided migration into the molecular layer, or from being pushed along by subsequently migrating neurons.
In the accompanying paper (17), evidence of a break in the external glial limiting membrane associated with molecular layer ectopias also supports the role of direct injury. Because of the association with autoimmunity both in animals and humans, it is tempting to suggest an immunopathogenic factor causing the injury, but no support for this hypothesis is as yet available. Whether injury is the only method for inducing neocortical ectopias, or whether there are other possible pathogenetic mechanisms for cerebrocortical microdysgenesis, remains an open question.
ACKNOWLEDGMENTS
This work was supported by NIH Grant HD 20806 and the Research Division of the Orton Dyslexia Society.